










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) vol.76 no.6 Ciudad Autónoma de Buenos Aires dic. 2016
ARTÍCULO ESPECIAL
Regulación de expresión de genes de la familia de β-globina humana, útil en la búsqueda de nuevos blancos terapéuticos para tratamiento de hemoglobinopatías
Karen G. Scheps1, 2, Viviana Varela1, 2
1Cátedra de Genética, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires,
2INIGEM (Instituto de Inmunología, Genética y Metabolismo), CONICET- Universidad de Buenos Aires, Argentina
Dirección postal: Dra. Karen G. Scheps, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, Junin 956, 1113 Buenos Aires, Argentina
e-mail: karenscheps@gmail.com
Recibido: 6-X-2015
Aceptado: 17-III-2016
Resumen
Durante la etapa embrionaria, el desarrollo fetal y la vida posnatal se expresan isoformas funcionalmente distintas de hemoglobina, producto de la combinación de cadenas polipeptídicas sintetizadas a partir de los distintos genes que componen las familias de α- y β-globina. En función de que la presencia de altos niveles de hemoglobina fetal (Hb F) es beneficiosa en síndromes falciformes y talasémicos graves, se plantea revisar las bases de la regulación de la expresión de los genes de la familia de β-globina, en particular los genes que codifican las cadenas de γ-globina (HBG1 y HBG2). En este trabajo se revisan los conocimientos sobre factores de transcripción y reguladores epigenéticos que gobiernan los eventos de encendido y apagado de los genes de la familia de β-globina. Se espera que la consolidación de estos conocimientos permita hallar nuevos blancos terapéuticos para el tratamiento de hemoglobinopatías.
Palabras clave: Hb F; Cluster de β-globina; Genética; Epigenética.
Abstract
Regulation of the β-globin gene family expression, useful in the search for new therapeutic targets for hemoglobinopathies
Different hemoglobin isoforms are expressed during the embryonic, fetal and postnatal stages. They are formed by combination of polypeptide chains synthesized from the α- and β- globin gene clusters. Based on the fact that the presence of high hemoglobin F levels is beneficial in both sickle cell disease and severe thalassemic syndromes, a revision of the regulation of the β-globin cluster expression is proposed, especially regarding the genes encoding the γ-globin chains (HBG1 and HBG2). In this review we describe the current knowledge about transcription factors and epigenetic regulators involved in the switches of the β-globin cluster. It is expected that the consolidation of knowledge in this field will allow finding new therapeutic targets for the treatment of hemoglobinopathies.
Key words: Hb F; β-globin cluster; Genetics; Epigenetics.
La producción de hemoglobina fetal (Hb F) es característica del período embrionario del desarrollo de un individuo. Sin embargo, pueden existir niveles elevados de Hb F (superior al 2.0% de la Hb circulante) en distintas situaciones de la vida extrauterina. Está comprobado que tanto en los síndromes falciformes como en las formas β-talasémicas graves es beneficiosa la presencia de niveles aumentados de Hb F. Por un lado, la Hb F posee un efecto antisickling al disminuir la polimerización de la Hb S, otorgando protección frente a las crisis de dolor, osteonecrosis, retinopatía proliferativa, úlceras en las piernas, secuestro esplénico y retraso en la menarca, disminuyendo las muertes en el período perinatal y aumentando la supervivencia1. Por otra parte, niveles elevados de Hb F atenúan el fenotipo clínico en síndromes talasémicos graves al disminuir el exceso de cadenas de α-globina libres2.
En función de estos hallazgos, se fomentó el interés por conocer las bases moleculares involucradas en la regulación de la expresión de los genes que codifican para cadenas de globina, con el fin de poder desarrollar terapias farmacológicas y, eventualmente génicas, cada vez más precisas. En forma empírica, por sus efectos sobre los niveles de Hb F, se instituyó la administración de hidroxiurea como tratamiento en pacientes con síndromes falciformes, aunque existe una gran variabilidad de respuesta en los distintos pacientes. En consecuencia, la meta es mejorar la calidad de los tratamientos, poniendo el foco en la regulación de la expresión de los genes de globina3, 4. Este trabajo tiene como objetivo actualizar los conocimientos en este campo.
Organización estructural de las familias de genes de las globinas
Los genes que codifican las cadenas de tipo α- y β-globina están organizados en familias génicas (o clusters).
La familia de α-globina mapea en la región subtelomérica del brazo corto del cromosoma 16 (16p13.3); contiene 3 genes que producen polipéptidos de 141 aminoácidos: HBZ, HBA2 y HBA1, 2 genes que se transcriben, aunque los productos de traducción no fueron detectados en humanos (HBM y HBQ1) y 2 pseudogenes (HBZP y HBAP1). El orden en que se encuentran los miembros de esta familia génica es, en dirección 5’ a 3’, HBZ-HBZP-HBM-HBAP1-HBA2-HBA1-HBQ1 (Fig. 1). La expresión en altos niveles de estos genes es guiada por un locus regulatorio altamente conservado entre especies (Multispecies Conserved Sequences, MCS), ubicado corriente arriba al cluster.
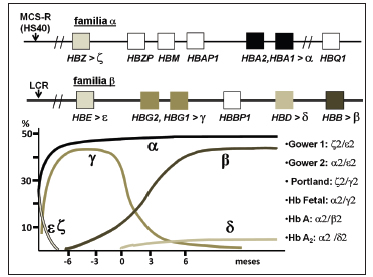
Fig. 1. Esquema de las familias de genes de α- y β-globina con sus regiones regulatorias y su expresión secuencial durante el desarrollo. Se indican las isoformas de Hb resultantes de las combinaciones de las distintas cadenas de tipo α- y β-globina.
Los genes que codifican las cadenas de tipo β-globina,se detallarán a continuación.
Organización del cluster de β-globina
La familia de genes de β-globina humana, se ubica en el brazo corto del cromosoma 11 (11p15.5), en una región pobre en genes, sin islas CpG asociadas, con un contenido de GC cercano al 40%. Se compone del gen ε, de expresión embrionaria (HBE1), 2 genes que codifican las cadenas de γ-globina, de expresión fetal: Gγ (HBG2) y Aγ (HBG1), 2 genes que codifican las cadenas tipo β del adulto: δ-globina (HBD) y β-globina (HBB) y un pseudogen de β-globina: ψβ (HBBP). El orden de los genes en el cluster es, en dirección 5’ a 3’, HBE1-HBG2-HBG1-HBBP-HBD-HBB (Fig. 1).
La expresión de los genes del cluster está regulada por elementos reguladores en cis (en el mismo cromosoma), proximales y distales. Los genes presentan motivos clásicos en las regiones promotoras como la caja TATA y 2 motivos específicos de genes de globina: caja CCAAT y caja CACCC5. Estos elementos se pueden encontrar en distinto orden y en distinto número de repeticiones en los diferentes genes del cluster. Adicionalmente, pueden encontrarse otros elementos activadores o silenciadores específicos en los distintos promotores.
Existe una transcripción basal de los genes de esta familia a partir de sus promotores. No obstante, para que haya alta eficiencia transcripcional, es necesaria la presencia de un elemento regulador distal, el locus de la región de control (Locus Control Region, LCR), que se ubica aproximadamente de 6 a 20 kb corriente arriba de HBE1.
El locus LCR está constituido por 5 sitios eritroide-específicos, HS1 a HS5 (de 250 a 500 pb) que presentan hipersensibilidad a DNAsaI (característico de regiones enhancer) y contienen secuencias blanco tanto para factores de transcripción ubicuos como específicos de la serie roja. Los sitios HS del LCR actúan en conjunto como un holocomplejo6, aunque la actividad enhancer se concentra en HS2, HS3 y HS4. HS5 funciona como aislante, al unir la proteína CTCF (CCCTC-Binding factor), que posee actividad de insulator o aislante, y evita la propagación de la activación, por parte del LCR a otras regiones de la cromatina.
Expresión temporal de las distintas hemoglobinas
El proceso eritropoyético, experimenta modificaciones durante le ontogenia: entre la segunda y la tercera semana de vida, en el saco vitelino, a partir de células mesodérmicas, aparecen eritrocitos primitivos, que presentan mayor tamaño que los definitivos y que ingresan a circulación como células nucleadas, alrededor del día 21 del desarrollo. Alrededor del día 23, comienza la colonización hepática por parte de células precursoras, producidas también extraembrionariamente, en el saco vitelino, capaces de dar lugar a células eritroides definitivas, a megacariocitos y a distintos granulocitos. A partir de esta colonización hepática, comienza la diferenciación de eritrocitos definitivos, convirtiéndose en el principal órgano productor de células rojas desde la décima semana del desarrollo hasta la vigésima cuarta. Entre el quinto y el sexto mes de vida, comienza a disminuir la producción de células sanguíneas en hígado y en órganos hematopoyéticos secundarios como el bazo y, en un proceso cortisol-dependiente, la médula ósea pasa a ser el órgano hematopoyético central.
Estos procesos de maduración del eritrocito y migración del órgano hematopoyético principal, se acompañan de cambios moleculares en los eritrocitos. La expresión de los distintos genes de los clusters de α- y β-globina está regulada a lo largo del desarrollo ontogénico de un individuo. Durante la etapa embrionaria, el desarrollo fetal y la vida posnatal, se sintetizan distintas isoformas de Hbs en las células eritroides. Todas comparten la capacidad de transporte de O2 y otros ligandos pero, a su vez, presentan características particulares, como diferencias en la afinidad por el O2 (Fig. 1).
En el período embrionario, en los eritrocitos primitivos, se expresan predominantemente el gen HBZ, del cluster de α-globina, que codifica para las cadenas de ζ-globina y el gen HBE1 del cluster de β-globina, que codifica para las cadenas de ε-globina, y ambas cadenas se combinan formando la isoforma Hb Gower 1. Esta forma es la de predominante circulación en las semanas 4 a 5 del desarrollo embrionario. En las semanas 6 a 7 se sigue encontrando en circulación Hb Gower 1, pero es mayoritaria la Hb Gower 2, que se compone de 2 cadenas de α-globina asociadas a 2 cadenas de ε-globina. Coincidentemente, cambia también el estadio madurativo del eritrocito primitivo en la circulación embrionaria, predominando los eritroblastos ortocromáticos.
La expresión de los genes embrionarios disminuye gradualmente al convertirse el hígado fetal en el principal sitio de maduración eritropoyética. Por un lado, se consolida la expresión de los genes HBA2 y HBA1 que codifican para las cadenas de α-globina, y por otro, aumenta la transcripción de los genes HBG2 y HBG1 que codifican las cadenas de γ-globina, que se expresan de forma mayoritaria hasta el período perinatal. La combinación de las cadenas de α-globina con las de γ-globina da lugar a la Hb F. En la transición del período embrionario al fetal pueden encontrarse otras Hbs producto de combinaciones entre las cadenas de expresión fetal y embrionaria, como la ya mencionada Hb Gower 2 (α2ε2) y la Hb Portland (ζ2γ2).
Alrededor del momento del nacimiento decae la expresión de cadenas de γ-globina y comienzan a expresarse los genes HBB y HBD, que codifican para cadenas de β- y δ-globina, respectivamente. La combinación de las cadenas de α-globina con las cadenas β da lugar a la Hb A, la fracción mayoritaria (cerca del 97% de Hb total circulante) aproximadamente a partir de los 6 meses de vida. La combinación de las cadenas de α-globina con las cadenas δ genera la Hb A2, que, en condiciones fisiológicas, representa un 2 a 3% de la Hb total circulante de la vida extrauterina. El porcentaje restante corresponde a la expresión residual de los genes de γ-globina, con la consecuente formación de Hb F, con valores fisiológicos inferiores al 2%.
Regulación de la expresión y eventos de "apagado" y "encendido" génico (switch) en el cluster de β-globina
Si bien los genes de este cluster fueron estudiados extensivamente (HBB fue uno de los primeros genes humanos en ser clonado10) constantemente surgen nuevos hallazgos sobre cómo se regula su expresión.
Ancestralmente, hasta después de la divergencia de secuencias en el ADN entre anfibios y amniotos, los clusters de α- y β-globina se encontraban organizados en tándem. De hecho, algunos marsupiales aún conservan genes huérfanos tipo-β en el cluster de α-globina11.
La transposición durante la evolución, de los genes de la familia de β-globina a una región genómica de expresión no constitutiva (dentro del cluster de receptores olfatorios), implicó la necesidad de cambios a nivel de la estructura de la cromatina, para que estos genes se expresaran con alta eficiencia: los promotores debieron demetilarse, se intercambiaron marcas en la cromatina y el cluster se relocalizó en su disposición nuclear desde una región de heterocromatina pericentromérica a zonas transcripcionalmente activas.
Se postula que la expresión de los genes del cluster de β-globina en células eritroides se produce por la interacción física entre las secuencias regulatorias (HS) del LCR y las secuencias promotoras del gen que se expresan a través de la formación de un bucle (loop), que excluye la cromatina intermedia. Se forma el Active Chromatine Hub (ACH), un compartimento celular específico, que determina la transcripción eficiente, mediada por la ARN polimerasa II6. La formación de esta estructura es necesaria para el reclutamiento de enzimas remodeladoras de la cromatina, para un eficiente ensamblado del complejo de pre-iniciación de la transcripción (PIC) así como para la estabilización del complejo intermediario para la reiniciación de la transcripción y la relocalización de los genes a fábricas transcripcionales, donde se concentran los factores transcripcionales y la ARN Polimerasa II.
En progenitores eritropoyéticos tempranos, un complejo pentamérico integrado por los factores de transcripción GATA2, TAL1, E2A, LMO2 y LDB1 se une a las secuencias blanco en HS2 de la región regulatoria y de las regiones promotoras del gen del cluster que debe expresarse según el desarrollo ontogénico, siendo GATA2 y TAL1 los puntos de contacto entre ambas regiones.
A medida que avanza la diferenciación de la serie roja, se produce un intercambio de los factores GATA: GATA2 es reemplazado por el factor maestro eritroide GATA1 y el complejo pentamérico se une al promotor del gen que se va a expresar. LDB1 es capaz de dimerizar (y multimerizar) por su dominio N-terminal, permitiendo la interacción de ambas regiones en cis y la formación del bucle (Fig. 2).
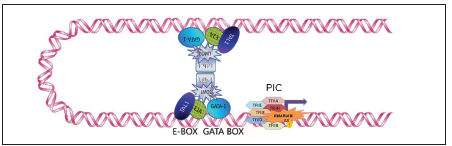
Fig. 2. Reclutamiento del complejo pentamérico y del complejo de pre-iniciación de la transcripción (PIC) en el cluster de β-globina. GATA1 y el heterodímero de TAL1-E2A se unen a sus secuencias blanco en ADN en el locus LCR y en los promotores de los genes del cluster. LMO2 actúa como puente, asociando GATA1 al heterodímero e interactúa también con LDB1. LDB1 es capaz de dimerizar y multimerizar permitiendo el acercamiento de los complejos montados en las distintas secuencias regulatorias. A partir de estas plataformas, GATA1 y TAL1 son capaces de reclutar proteínas remodeladoras de la cromatina y otros factores transcripcionales, permitiendo la formación del PIC (ARN Polimerasa II y factores generales de transcripción -TF-) y una transcripción eficiente por parte de la ARN Polimerasa II.
Como se mencionó previamente, en el cluster de β-globina se dan 2 eventos de "apagado" y "prendido" de genes (switch) durante el desarrollo. La activación de la transcripción del gen situado más 5’ en el cluster y más próximo al LCR (HBE1), implica la formación del primer complejo de activación, en el período embrionario. Se cree que la activación, de este gen está mediada por un mecanismo de tracking (migración de los factores transcripcionales) además de la formación del bucle. Desde el LCR se reclutarían las enzimas remodeladoras de la cromatina y este locus actuaría como plataforma de montaje del complejo de preiniciación de la transcripción (PIC), transmitiendo la activación a través de la cromatina por medio de la ARN polimerasa II.
Es notable que el silenciamiento de HBE1 no se da por competencia con otros genes por el LCR, sino que es autónomo: depende de la interacción de factores represores con una secuencia silenciadora ubicada corriente arriba a HBE1, donde se encuentran sitios de unión superpuestos para GATA1 e YY1 (Yin Yang 1, un factor transcripcional de tipo proteína dedo de zinc GLI-Kruppel que actúa principalmente como represor de la transcripción), que contribuyen al "apagado", y 2 motivos a los que se unen el complejo DRED (Direct Repeat Erythroid-Definitive, formado por los receptores nucleares huérfanos TR2 y TR4) y COUP-TFII (COUP transcription factor 2 o NR2F2)12, 13.
Por último, la metilación del gen juega un rol importante en el mantenimiento de su silenciamiento y se encontraría mediada indirectamente por MDB2 (Methyl-CpG binding domain protein 2), que se une a citosinas metiladas por fuera del gen y reclutaría otras proteínas efectoras14, posiblemente DNMT1 (DNA (cytosine-5-)-methyltransferase 1).
En paralelo al "apagado" de HBE1, se produce el "encendido" de los genes que codifican para las cadenas de γ-globina que presentan sitios de unión de factores transactivadores específicos del período fetal en las regiones promotoras, las cuales estimulan su transcripción. Para la expresión de estos genes sería esencial la actividad de factores Krüper-like (aparentemente KLF2, 5 y 13)15.
Por otra parte, en la activación de HBG2 y HBG1, también estaría involucrada la unión del complejo Stage Selector Protein (SSP), formado por el heterodímero de TFCP2 y NF-E4, al motivo Stage Selector Element (SSE) en el promotor de estos genes.
Es interesante que solo algunos primates del viejo mundo adquirieron expresión diferencial de genes en los períodos embrionario y fetal16. Asimismo, junto con el patrón de expresión fetal de los genes de γ-globina durante la evolución de los primates, los promotores de estos genes adquirieron una mayor concentración local de residuos CpG (5 CpG en las primeras 105 pb), ausentes en otros promotores del cluster.
Numerosos factores transcripcionales y complejos multiproteicos cooperan en el silenciamiento de los genes de γ-globina, alterando la estructura de la cromatina (detallado más adelante) y facilitando su metilación, entre ellos, los factores capaces de unirse a las repeticiones directas (DR1) en las regiones promotoras de estos genes (DRED y COUP-TFII), el complejo NuRD, que contiene desacetilasas de histonas y es reclutado mediante la interacción de GATA1 con FOG1, -las enzimas DNMT3A (DNA (cytosine-5)-methyltransferase 3A, responsable de la metilación de estos genes), PRMT5 (Protein arginine N-methyltransferase 5, involucrada en la metilación de residuos de arginina en las histonas)17 y FOP1, una proteína asociada a la cromatina, que ejercería este efecto regulando los niveles de SOX6.
La región intergénica entre HBG1 y HBD participa en el silenciamiento de los genes de expresión fetal: contiene un tracto polipirimidina de 250 pb al que se une por medio del factor IKZF1 (IKAROS family zinc finger 1), el complejo PYR (que nuclea a componentes de los complejos NuRD y SWI/SNF) y además contiene sitios de unión para BCL11A, una proteína dedo de zinc tipo C2H2 (dominio característico de unión al ADN). Recientemente se descubrió el rol de esta proteína (asociada previamente a la patogenia de linfomas) en la atenuación de la expresión de los genes de γ-globina, a partir de estudios de Genome Wide Association (GWAS) que asociaron polimorfismos en este gen con cambios en los niveles de Hb F, tanto en población normal como en pacientes con distintas hemoglobinopatías18. El gen BCL11A comienza a expresarse en eritrocitos definitivos, siendo su principal inductor el factor maestro eritroide KLF1. En el momento del segundo switch del cluster de β-globina se expresan principalmente dos isoformas que difieren en el uso del exón 3’ terminal: L y XL (de mayor peso molecular), siendo esta última la más abundante y la que ocupa mayormente el cluster de β-globina. BCL11A se une tanto a la región intergénica entre HBG1 y HBD, como al LCR, promoviendo interacciones de largo alcance entre el locus regulatorio y el gen HBB, excluyendo a los genes HBG de su interacción con las secuencias del LCR. BCL11A también interviene en el silenciamiento de estos genes mediante el reclutamiento de distintos factores de transcripción: es capaz de asociarse con SOX6, que se une a los promotores de los genes HBG. SOX6 actúa como un "factor arquitecto", ya que se une al surco menor del ADN e induce curvaturas drásticas en la cromatina, reorganizando la estructura local y el acceso de los factores de transcripción asociados al ADN.
A su vez, BCL11A puede interactuar con un gran número de factores transcripcionales y efectores epigenéticos: FOG1, con el complejo NuRD, con IKZF1, con componentes del complejo remodelador de la cromatina SWI/SNF, con EZH2 del complejo Polycomb, con otros complejos co-represores como NCoR/SMRT, BCOR, el complejo desacetilasa SIN3, el complejo demetilasa de histonas LSD1/CoREST y con la ADN metil-transferasa DNMT1, implicada en el mantenimiento del silenciamiento de estos genes19. Recientemente se demostró la participación de 2 ARNs de interferencia, LIN28B y microRNA-486-3p20, en la regulación negativa de la expresión de BCL11A.
Los estudios de GWAS también revelaron la influencia de otro locus en la regulación de la expresión de los genes HBG: la región intergénica de los genes HBS1L y MYB, en el cromosoma 6q23.3. Variantes de secuencia en esta región se asociaron a un silenciamiento inefectivo de los genes fetales21. Distintas pruebas afirman que, en esta región, se encontrarían secuencias regulatorias de la expresión de MYB, un gen que codifica un factor hematopoyético esencial, el cual actúa como regulador maestro, ajustando la cinética de maduración eritroide: cumple un rol crítico en la coordinación de la expresión de los genes de globina, la regulación del ciclo celular y la diferenciación. El gen MYB se expresa en niveles altos en células inmaduras en proliferación, y sus niveles disminuyen a medida que avanza la diferenciación de la célula eritroide. Se halló al menos un mecanismo regulatorio postranscripcional, atenuador de su expresión: su ARNm es blanco de los micro-ARNs (miRNAs) miR-15a y miR-16a22. Estudios recientes demostraron que MYB regula positivamente la expresión de KLF1, que a su vez estimula la expresión de BCL11A y los factores KLF3 y KLF8, todos ellos implicados en el silenciamiento de HBG2 y HBG1. El factor MYB también influiría en el silenciamiento de los genes que codifican las cadenas de γ-globina, actuando como regulador de TR2 y TR4, los componentes del complejo DRED. A su vez, se postuló una acción cooperativa de MYB con DNMT1 en el silenciamiento de HBE123.
A medida que se atenúa la expresión de los genes HBG en el período perinatal, aumentan los niveles de transcripción de HBD y principalmente de HBB, que hasta ese momento del desarrollo se encontraban silenciados por metilación de sus promotores y por la competencia por la función de apertura de la cromatina por parte de los genes del cluster ubicados corriente arriba. El factor KLF1, además de estimular la transcripción de BCL11A, es esencial para la activación de HBB: estabiliza el loop entre el LCR y la región promotora de HBB, ejerce un rol fundamental en la traslocación del gen a las fábricas transcripcionales y además recluta al complejo remodelador de la cromatina E-RC1, relacionado al complejo SWI/SNF, produciendo la apertura de la cromatina en el promotor del gen. Para poder ejercer estas acciones, el factor KLF1 debe transactivarse por acetilación en la lisina 28824.
Otro factor regulatorio es NF-E2 que es un heterodímero constituido por los factores p18 (ubicuo) y p45 (eritroide-específico); se une a secuencias blanco en el LCR en las distintas etapas del desarrollo ontogénico; pero en el período perinatal se une además a la región promotora de HBB e interviene en la transferencia eficiente de la ARN Polimerasa II desde el LCR al promotor. Otras proteínas, asociadas a modificaciones epigenéticas, resultan fundamentales para la activación del gen HBB, como UTX (con actividad H3-K27 demetilasa), las acetiltransferasas de histonas PCAF (P300/CBP-associated factor) y KAT2A y la metilasa de histonas PRMT1 (Protein arginine N-methyltransferase 1).
Modificaciones en las marcas epigenéticas en el cluster de β-globina durante la ontogenia
Metilación del ADN
En células no eritroides, las regiones promotoras de los genes del cluster se encuentran metilados, mientras que en las células eritroides se observa hipometilación de los promotores en los dominios implicados en transcripción activa.
Modificaciones en las marcas de histonas
La región LCR, involucrada en la regulación de la transcripción del cluster, y los promotores de los genes, sufren modificaciones en las marcas de histonas propias de regiones enhancers y promotoras, respectivamente, y de acuerdo al estado de activación de los genes.
En células no eritroides y antes de comenzar la transcripción de los genes del cluster en células eritroides, las histonas asociadas al LCR presentan marcas represivas, como las dobles metilaciones (me2) en histona H3: H3K9me2, H3K36me2 y H3K27me2. Cuando empiezan a expresarse los genes de la familia de la β-globina, esas marcas son reemplazadas por las de activación como las acetilaciones (Ac) H3K9Ac, H3K36Ac y H3K27Ac, que se mantienen durante los distintos estadios del desarrollo. Esta región también presenta un patrón de modificaciones propio de regiones enhancers caracterizado por altos niveles de H3K4me1 y bajos niveles de H3K4me3.
Los promotores de los genes del cluster presentan distintas marcas a lo largo de la ontogenia: cuando se silencia HBE1 se encuentra la marca represiva H3K9me2 y están deplecionadas marcas características de activación, como la acetilación de la H3 y H3K4me3. Durante la transcripción activa de HBG2 y HBG1 se encuentran marcas como la acetilación de H3 (K9 y K27), H3K4me2 y H3K4me3, mientras que al sufrir silenciamiento se observa, al igual que en HBE1, presencia de H3K9me2 y pérdida de las marcas de activación17. En la activación de HBB se ve involucrada la metilación asimétrica de H4R3 por PRMT1, que promueve la adquisición de otras marcas activantes como H3K9Ac y H3K14Ac25. También se observó la marca H3K4me317.
Regulación de la expresión del cluster de β-globina como blanco terapéutico
Desde la década de 1970 se conocen los efectos beneficiosos de niveles aumentados de Hb F en pacientes con síndromes falciformes. En 1984 se documentaron los primeros casos de incrementos de esta isoforma de la Hb por efecto de la hidroxiurea y en 1998 la US Food and Drug Administration (FDA) aprobó el uso de esta droga para el tratamiento de adultos con anemia falciforme con crisis de dolor recurrentes moderadas a graves26. El uso de esta droga es uno de los tratamientos más difundidos para pacientes con síndromes falciformes. No obstante, en la práctica médica se observó variabilidad individual de respuesta al tratamiento y, por otra parte, la hidroxiurea no es efectiva para prevenir todas las complicaciones asociadas a estos síndromes. Poco se conoce de los mecanismos por los cuales esta droga ejerce su efecto; recientemente se demostró que es capaz de disminuir la expresión de BCL11A, KLF1 y TAL127. A partir de los avances en el conocimiento de los mecanismos fisiológicos que regulan la expresión, y en consecuencia, los niveles de Hb F, se están buscando nuevas alternativas para el tratamiento de los síndromes falciformes y los talasémicos graves. Los avances en el conocimiento de la regulación de la expresión de los genes de globina abre el camino hacia la "medicina personalizada" con el desarrollo de nuevos fármacos o nuevos usos de fármacos existentes, y por otro, da impulso a estrategias de terapia génica.
En función de los conocimientos sobre los mecanismos epigenéticos que regulan la expresión del cluster de β-globina, se están llevando a cabo ensayos clínicos –en distintas fases– con drogas aprobadas por la FDA capaces de incrementar los niveles de Hb F; por ejemplo, la decitabina, que es un inhibidor de la ADN metil transferasa DNMT1, para el tratamiento de pacientes con enfermedad de células falciformes (ClinicalTrials.gov Identificador: NCT01685515), que actualmente se encuentra en Fase 1. Estudios in vitro demostraron que el antidepresivo tranilcipromina, un inhibidor de la Lysine-specific histone demethylase 1A (LSD1), también induce la síntesis de Hb F en células progenitoras eritroides y que tiene un efecto sinérgico con decitabina28.
Muchos de los desarrollos actuales de terapia génica para el tratamiento de las hemoglobinopatías contemplan la transferencia del conocimiento básico de los mecanismos involucrados en la regulación de la expresión a desarrollos que permitan estimular la síntesis de Hb F.
Para ello, se están ensayando dos estrategias: por un lado, la activación de la expresión de HBG actuando sobre la región promotora. Se encuentra en desarrollo el uso de un vector lentiviral con una proteína de fusión de LDB –el factor que compone el complejo pentamérico y permite la interacción entre el LCR y las regiones promotoras– y una proteína con dominio dedo de zinc capaz de reconocer específicamente el promotor de los genes HBG. La otra estrategia para aumentar los niveles de Hb F se basa en la atenuación de la síntesis de los inhibidores de la expresión de los genes HBG como el uso de short interfering ARNs (siRNAs) y shorthairpin ARNs (shRNAs) para silenciar la expresión de BCL11A y KLF1 o promover la expresión de miRNAs endógenos cuyos blancos regulan negativamente la expresión de los genes que codifican las cadenas de γ-globina29, 30.
Se espera que la profundización de estos conocimientos pueda aplicarse en beneficio de los pacientes que padecen formas graves de hemoglobinopatías en un futuro cercano, al permitir la aplicación de medicina genómica y el desarrollo y utilización de fármacos capaces de atenuar el fenotipo.
Conflicto de intereses: Ninguno para declarar
1. Steinberg MH. Predicting clinical severity in sickle cell anaemia. Br J Haematol 2005; 129: 465-81. [ Links ]
2. Forget BG, Bunn HF. Classification of the disorders of hemoglobin. Cold Spring Harb Perspect Med 2013; 3: a011684. [ Links ]
3. Wienert B, Funnell AP, Norton LJ, et al. Editing the genome to introduce a beneficial naturally occurring mutation associated with increased fetal globin. Nat Commun 2015; 6: 7085. [ Links ]
4. Cui S, Lim KC, Shi L, et al. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice. Blood 2015; 126: 386-96. [ Links ]
5. Patrinos GP, Antonarakis SE. Human hemoglobin. En: Speicher M, Antonarakis SE., Motulsky AG, eds. Vogel and Motulsky’s Human Genetics. Berlin: Springer-Verlag Berlin Heidelberg, 2010, p 365-401.
6. Palstra RJ, de Laat W, Grosveld F. β‐globin regulation and long‐range interactions. Adv Genet 2008; 61: 107-42.
7. Higgs DR, Vernimmen D, Wood B. Long‐range regulation of α‐globin gene expression. Adv Genet 2008; 61: 143-73.
8. Coelho A, Picanço I, Seuanes F, Seixas MT, Faustino P. Novel large deletions in the human α-globin gene cluster: clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol Dis 2010; 45: 147-53.
9. Ribeiro DM, Sonati MF. Regulation of human alpha-globin gene expression and alpha-thalassemia. Genet Mol Res 2008; 7: 1045-53. [ Links ]
10. Wilson JT, Forget BG, Wilson LB, Weissman SM. Human globin messenger RNA: importance of cloning for structural analysis. Science 1977; 196: 200-2. [ Links ]
11. Hoffmann FG, Storz, JF, Gorr TA, Opazo JC. Lineage-specific patterns of functional diversification in the α-and β-globin gene families of tetrapod vertebrates. Mol Biol Evol 2010; 27: 1126-38.
12. Raich N, Clegg CH, Grofti J, Roméo PH, Stamatoyannopoulos G. GATA1 and YY1 are developmental repressors of the human epsilon-globin gene. EMBO J 1995; 14: 801-9. [ Links ]
13. Sankaran VG, Xu J, Orkin SH. Advances in the understanding of haemoglobin switching. Br J Haematol 2010; 149: 181-94. [ Links ]
14. Rupon JW, Wang SZ, Gnanapragasam M, Labropoulos S, Ginder GD. MBD2 contributes to developmental silencing of the human ε-globin gene. Blood Cells Mol Dis 2011; 46: 212-19.
15. Zhang P, Basu P, Redmond LC, et al. A functional screen for Krüppel-like factors that regulate the human gamma-globin gene through the CACCC promoter element. Blood Cells Mol Dis 2005; 35: 227-35. [ Links ]
16. Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Exp Hematol 2005; 33: 259-71. [ Links ]
17. Saunthararajah Y, Lavelle D, DeSimone J. Epigenetic regulation of globin genes and disturbances in hemoglobinopathies. En: Lübbert M, Jones, PA, eds. Epigenetic therapy of cancer. Berlin: Springer-Verlag Berlin Heidelberg, 2014, p 89-106. [ Links ]
18. Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 2007; 39: 1197-9. [ Links ]
19. Xu J, Bauer DE, Kerenyi MA, et al. Core pressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci USA 2013; 110: 6518-23. [ Links ]
20. Ginder GD. Epigenetic regulation of fetal globin gene expression in adult erythroid cells. Transl Res 2015; 165: 115-25. [ Links ]
21. Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA 2008; 105: 11869-74. [ Links ]
22. Sankaran VG, Menne TF, Šćepanović D, et al. MicroRNA-15a and-16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci USA 2011; 108: 1519-24.
23. Roosjen M, McColl B, Kao B, Gearing LJ, Blewitt ME, Vadolas J. Transcriptional regulators Myb and BCL11A interplay with DNA methyltransferase 1 in developmental silencing of embryonic and fetal β-like globin genes. FASEB J 2014; 28: 1610-20.
24. Sengupta T, Chen K, Milot E, Bieker JJ. Acetylation of EKLF is essential for epigenetic modification and transcriptional activation of the β-globin locus. Mol Cell Biol 2008; 28: 6160-70.
25. Li X, Hu X, Patel B, et al. H4R3 methylation facilitates β-globin transcription by regulating histone acetyltransferase binding and H3 acetylation. Blood 2010; 115: 2028-37.
26. Wong TE, Brandow AM, Lim W, Lottenberg R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood 2014; 124: 3850-7 [ Links ]
27. Grieco AJ, Billett HH, Green NS, Driscoll MC, Bouhassira EE. Variation in gamma-globin expression before and after induction with hydroxyurea associated with BCL11A, KLF1 and TAL1. PLoS One 2015; 10: e0129431. [ Links ]
28. Pace BS, Liu L, Li B, Makala LH. Cell signaling pathways involved in drug-mediated fetal hemoglobin induction: Strategies to treat sickle cell disease. Exp Biol Med (Maywood) 2015; 240: 1050-64. [ Links ]
29. Chandrakasan S, Malik P. Gene therapy for hemoglobinopathies: the state of the field and the future. Hematol Oncol Clin North Am 2014; 28: 199-216. [ Links ]
30. Finotti A, Breda L, Lederer CW, et al. Recent trends in the gene therapy of β-thalassemia. J Blood Med 2015; 6: 69-85.

