











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La atrofia muscular espinal (AME) es una enfermedad neuromuscular que afecta aproximadamente a 1 entre 6000 a 11 000 nacidos vivos1-4 con una frecuencia de portadores de hasta 1 en 60. Es considerada la segunda causa de muerte por enfermedad autosómica recesiva, detrás de la fibrosis quística.3,4 Es causada por mutaciones en el gen de supervivencia de la neurona motora 1 (SMN1), ubicado en el brazo largo del cromosoma 5 (locus 5q13.2), que conllevan la degeneración de las neuronas motoras de la médula espinal, produciendo atrofia y debilidad muscular progresiva. Otro gen, el SMN2, codifica una proteína similar a la producida por el SMN1, pero en menor cantidad; compensa de alguna manera la pérdida del gen SMN1 y hace que la gravedad del cuadro sea menor.3'5-8
El mayor número de copias del SMN2 se suele correlacionar con un fenotipo menos grave.
La AME está clasificada en cuatro tipos (0 a IV) según la edad de aparición de los síntomas y la gravedad. Nos ocuparemos de las formas I, II y III. Aproximadamente el 50 % de los pacientes son de tipo I, el más grave y de inicio más temprano. No sobreviven más allá de los 2 años de vida, no logran la sedestación y presentan dificultades para deglutir, alimentarse y respirar.8-10
En AME tipo II los síntomas aparecen más tarde. Los niños pueden sentarse sin apoyo y algunos logran pararse (con apoyo), pero nunca caminar de forma independiente. Pueden presentar debilidad al deglutir o masticar, y dificultad respiratoria. La tasa de supervivencia es más alta que la del tipo I.4'8
Los pacientes con AME III logran caminar, pero a menudo pierden la posibilidad de hacerlo a medida que avanza la enfermedad.4,8
Los signos clínicos en AME pueden variar ampliamente; algunos, como la hipotonía y el retraso motor, son comunes a otras patologías neuromusculares.11 Aunque el reconocimiento de la AME está aumentando, el retraso en el diagnóstico es frecuente y la primera alerta depende de la observación y el reconocimiento de los primeros signos.
En los últimos años se han desarrollado terapéuticas que cambiaron el paradigma que definía a la AME como una enfermedad sin tratamiento.12 La primera terapia transformadora para la AME,13 que utiliza la modificación de la transcripción del ARN del SMN2, fue aprobada por la Food and Drug Administration (FDA) en diciembre de 2016 para todas las edades; y la segunda,14 el reemplazo del gen SMN1, fue aprobada en mayo de 2019 para niños menores de 2 años. Ambas están cambiando la historia natural de la enfermedad. Un tercer tratamiento con una pequeña molécula capaz de modificar el sitio de splicing del SMN2 ha sido recientemente aprobado por la FDA.15
Este estudio intenta reconstruir el camino al diagnóstico recorrido por pacientes con AME identificando hitos como la edad de aparición de los primeros signos, momento de la sospecha clínica y edad a la confirmación diagnóstica, así como el tiempo transcurrido entre cada una de estas. El estudio permitió analizar algunas de las causas de la demora diagnóstica.
POBLACIÓN Y MÉTODOS
Se realizó un estudio observacional de corte transversal en 112 pacientes que pudieron ser contactados (convenience sampling) con diagnóstico de AME tipo I, II y III entre noviembre de 2020 y septiembre de 2021 en Argentina. Se incluyeron 40 pacientes con AME I, 48 pacientes con AME II y 24 pacientes con AME III.
Los criterios de inclusión para poder participar del estudio fueron, por un lado, que el paciente tuviera un diagnóstico genético confirmado con mutaciones en el gen SMN1 (incluidas deleciones, duplicaciones y mutaciones puntuales) y que el paciente, un familiar y/o un cuidador fueran capaces de responder la entrevista. El tiempo desde el diagnóstico no fue un factor condicionante a la hora de ser incluidos. Dentro de los criterios de exclusión, el paciente no tenía que tener otro familiar diagnosticado antes que él/ella que hubiese facilitado su diagnóstico.
Neurólogos especializados en enfermedades neuromusculares efectuaron entrevistas telefónicas o presenciales con formato de interrogatorio al paciente y/o a sus familiares. Estas se complementaron con la revisión de las historias clínicas toda vez que se planteara una duda o contradicción sobre el dato recabado. Los padres y/o familiares o el propio paciente, según el caso, fueron informados de los objetivos del estudio. Las entrevistas fueron únicas o divididas hasta un número máximo de 4 según la determinación del entrevistador. No se utilizó una guía preestablecida.
El interrogatorio se adecuó en cada caso hasta obtener la información necesaria para recabar los siguientes datos: primer/os síntoma/s (o signo/s) relacionados con la enfermedad, momento de aparición y quién/quiénes lo/s notó/notaron. Primer profesional consultado (médico especialista u otro), diagnósticos dados, derivación a otros especialistas y momento en que ocurrió, edad de sospecha clínica de diagnóstico de AME, momento de la confirmación diagnóstica mediante estudio molecular, especialidad que realizó el diagnóstico y otros estudios solicitados.
El estudio fue aprobado por un comité de ética. La información obtenida se manejó en forma confidencial. Se tomaron todas las precauciones necesarias para proteger la privacidad y la confidencialidad de la información de los participantes del estudio.
Los datos fueron analizados utilizando estadísticas descriptivas. Las variables continuas fueron informadas como mediana (Me), rango intercuartílico (RIC) y rango (R), mientras que las categóricas fueron informadas como porcentaje (%).
RESULTADOS
Datos demográficos
La muestra estuvo formada por 112 pacientes (el 45 % de sexo femenino), de los cuales 40 presentan AME tipo I; 48, AME II y 24, AME III.
El rango de edades de los pacientes al momento de la encuesta fue de 10 meses a 38 años.
Primeros signos y su reconocimiento
En la Tabla 1 se exponen, para cada tipo de AME, los datos referidos a las edades en las que se observó el primer signo, realización de la primera consulta, momento de la sospecha clínica y confirmación del diagnóstico mediante la prueba molecular. Asimismo, se detallan los tiempos transcurridos entre el primer signo detectado y la primera consulta realizada, entre la primera consulta y el momento de la sospecha clínica de AME, entre la sospecha clínica y el diagnóstico confirmado. Finalmente, se muestra el tiempo que medió entre el reconocimiento del primer signo y el diagnóstico confirmado.
Si se tiene en cuenta la mayor disponibilidad de las pruebas moleculares diagnósticas en el país en los últimos años, y tomando el año 2018 como referencia, en AME I, el tiempo al diagnóstico confirmado desde los primeros signos fue más breve para aquellos pacientes nacidos entre 2018 y 2020 que para los nacidos entre 2005 y 2018. Para el primer grupo, la mediana (Me) fue de 1 mes (m) (RIC: 1-3; R: 0-11 m), mientras que, para el grupo de niños mayores, la Me fue de 2,5 m (RIC: 1-4,3; R: 0-10 m).
Los primeros signos clínicos detectados tanto por médicos como familiares se resumen en la Tabla 2.

Tabla 1: Eventos cronológicos expresados en meses
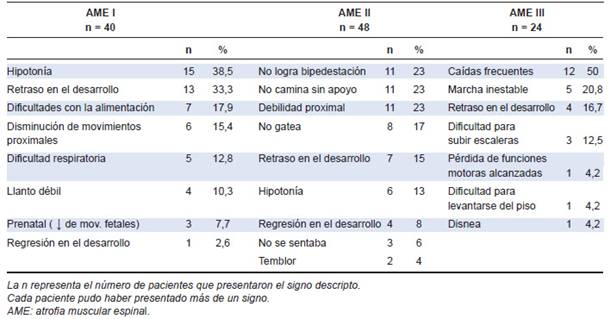
Tabla 2: Primer signo detectado

TABLA 3: Diagnósticos comunicados a la familia previos a la sospecha clínica de AME AME I AME II AME III
La detección de los primeros signos en AME I fue realizada por los padres en el 72,5 % de los casos (n = 29), por el neonatólogo en el 5 % (n = 2), por el pediatra en el 12,5 % (n = 5), por los abuelos en el 7,5 % (n = 3) y por un equipo multidisciplinara EM, grupo de médicos que intervinieron en forma conjunta durante una internación o emergencia sin poder ientificar qué especialista solicitó las pruebas diganósticas) en el 2,5 % (n = 1).
En AME II los signos fueron detectados en el 85 % de los casos por los padres (n = 41), por los abuelos en el 10,4 % (n = 5) y por tíos en el
4,2 % (n = 2).
En AME III los padres detectaron los signos el 75 % de las veces (n = 18); el pediatra, el 4,2 % (n = 1); un/a maestro/a, el 8,3 % (n = 2); los abuelos, tío o el propio paciente, el 4,2 % (n = 1) en cada caso.
De un total de 72 pacientes (48 AME II + 24 AME III) en los que los controles médicos rutinarios son menos frecuentes que en los primeros meses de vida, solo un médico pudo advertir los primeros signos de la enfermedad.
El especialista más frecuentemente visitado en la primera consulta fue el pediatra (82,5 %, n = 33
en AME I; 95,8 %, n = 46 en AME II, y 66,6 %, n = 16 en AME III). Los otros especialistas visitados se observan en la Figura 1.
En la Tabla 3se resumen los diagnósticos comunicados a la familia previos a la sospecha clínica de AME.
La mediana de consultas realizadas hasta la sospecha clínica fue para AME I: 3 (RIC 2-3, R: 1-6), AME II: 4 (RIC 3-4, R: 2-15) y AME III: 4 (RIC 3-5, R: 2-11).
El número de pacientes que consultaron a las distintas especialidades hasta la sospecha diagnóstica fueron en AME I (n = 40): pediatría 39, neurología 33, EM 7, genética clínica 4, neumonología 3 y obstetricia 1. En AME II (n = 48): pediatría 44, neurología 43, traumatología 11, genética clínica 7, endocrinología 2, otorrinolaringología 1, fisiatría 1, oftalmología 1 y kinesiología 1. En AME III (n = 24): neurología 22, pediatría 17, traumatología 17, fisiatría 2, kinesiología 2, reumatología 1, neurocirugía 1.
El estudio molecular diagnóstico fue solicitado en AME I por el neurólogo en el 82,5 % de los casos (n = 33), por el EM en el 15 % (n = 6) y por el genetista en el 2,5 % (n = 1). En AME II, por el neurólogo en el 95,8 % (n = 46) y por el genetista en el 4,2 % (n = 2). En AME III por el neurólogo en el 91,7 % de los casos (n = 22), y por un neuroortopedista y un genetista 4,2 %, (n = 1) en ambos casos.
Los estudios complementarios solicitados incluyeron electromiografía, ecografía cerebral, resonancia magnética nuclear (RMN) de cerebro, radiografía de tórax/caderas, tomografía de cerebro, creatinina-cinasa (CPK), biopsia muscular/nervio, toxina botulínica, determinación de alfa-glucosidasa, videodeglución, RMN de columna, cariotipo, electroencefalograma y punción lumbar.
El estudio genético se solicitó de manera inicial en 9 pacientes AME I (22,5 %), en 8 AME II (16,7 %) y en ninguno en AME III.
DISCUSIÓN
La AME es una enfermedad neuromuscular con una alta morbimortalidad, particularmente en las formas I y II. Conlleva graves complicaciones y requiere medidas de atención y apoyo tempranas que se pueden encontrar en diferentes guías de tratamiento.16-18
La progresiva disminución de motoneuronas compensada parcialmente con mecanismos reinervatorios se correlaciona con la aparición de los signos clínicos.19
Las nuevas terapias basadas en oligo nucleótidos antisentido, transferencia génica y moléculas modificadoras del splicing están mejorando el pronóstico y el curso evolutivo de la enfermedad.12-15 Cuanto antes se instalen, mayor será el beneficio terapéutico. El diagnóstico precoz resulta fundamental para favorecer el pronóstico de los pacientes.20
Una revisión sistemática de la literatura acerca de la demora diagnóstica en AME, reveló solo en 11 trabajos información sobre la edad de inicio, mientras que en 5 se informó la edad de inicio y la del diagnóstico confirmado, y se concluye que la demora diagnóstica es común en AME.21
Un estudio reciente en 5 centros italianos con un número importante de pacientes reveló también un retraso significativo en el diagnóstico de AME.22
Nuestro estudio es el primero de este tipo en Argentina y provee datos respecto de la edad de aparición de los primeros signos y de las demoras en el diagnóstico. Además, proporciona datos adicionales que permiten analizar sus posibles causas.
Los resultados muestran que existe un retraso importante entre la aparición de los primeros signos y el diagnóstico definitivo. Se puede observar que no hubo demoras entre la observación del primer signo y la primera consulta. Sin embargo, las diferencias entre la observación del primer signo y la confirmación diagnóstica fueron progresivas: menores en AME I, intermedias en AME II y máximas en AME III, lo cual se correlaciona con la gravedad y la rápida evolución de la enfermedad.
La causa de la demora en el diagnóstico se debe fundamentalmente a la falta de sospecha clínica por parte del médico interviniente, que muchas veces desestima o malinterpreta los signos referidos por los padres, según lo reflejan los diagnósticos alternativos invocados.
También existen retrasos significativos entre la sospecha clínica y el diagnóstico confirmado, especialmente en las formas II y III. En AME I, donde la presentación es más precoz y llamativa, el proceso diagnóstico resulta más acelerado. Asimismo, juegan su papel la organización del sistema de salud y la accesibilidad a los estudios moleculares, ahora más disponibles. En los últimos años, este hecho, asociado a la mayor concientización respecto de la enfermedad, ha permitido el acortamiento de los tiempos diagnósticos, como puede constatarse cuando se divide a la población de AME I en nacidos antes o después de 2018.
Ante la falta de sospecha clínica, factor más importante en la demora diagnóstica, no se realizaron en forma oportuna y temprana las correspondientes derivaciones a los especialistas en neurología, que resultaron los solicitantes del estudio genético en el 90 % de los casos.
Los estudios complementarios denotan que un número significativo de ellos no está orientado hacia el diagnóstico de AME ni de otras enfermedades neuromusculares, sino hacia patologías de origen central u ortopédico.
El número de médicos de cabecera a cargo de los controles que advirtieron a la familia sobre el primer signo en las tres formas de AME fue de 6 para el total de los pacientes (n = 112), lo que representó el 5,3 % de los casos.
Deben también considerarse factores culturales, como la errónea percepción de que los trastornos motores son de resorte traumatológico u ortopédico, y no neurológico.
Podemos concluir que, de modo similar a otras regiones en el mundo, aún existe demora diagnóstica en la AME que es atribuible en gran medida a la falta de sospecha clínica de la enfermedad. Acortar los tiempos de diagnóstico resulta fundamental para el éxito de las terapias actuales.
Los programas de detección de AME en recién nacidos tienen un gran potencial para identificar niños afectados en una etapa asintomática, lo que permitiría el inicio de la terapia antes de que ocurra un mayor daño de las neuronas motoras.23
Mientras tanto, es de particular relevancia la concientización de los médicos que brindan atención primaria a los pacientes de estas edades a través de programas de educación médica en los que se ponga foco en el reconocimiento y diagnóstico temprano de esta y de otras enfermedades neuromusculares.
Conflicto de intereses: En lo relativo a este trabajo, los Dres. Dubrovsky, Mesa y Vázquez recibieron honorarios por participación en Consejos Asesores de Novartis.
Otros potenciales conflictos de intereses en relación con la industria farmacéutica se mencionan a continuación:
Dr. Chloca, Dra. Morosini, Dra. Bolano, Dr. Jáuregui, Dr. Flores: sin conflicto de intereses.
Dra. Mesa: ha recibido honorarios por asesoramiento científico de los laboratorios PTC, Sarepta, Biogen, Avexis, Novartis, Roche. Ha recibido honorarios por conferencias de algunas de las industrias mencionadas. Dr. Vazquez: ha recibido honorarios por asesoramiento científico de los laboratorios Biogen, Avexis, Novartis, Roche. Ha recibido beca para investigación de Novartis y actividades académicas de Biogen. Ha recibido honorarios por conferencias de algunas de las industrias mencionadas, así como de PTC y Sarepta.
Dra. Pirra: ha recibido honorarios por asesoramiento científico de los laboratorios PTC, Sanofi Genzyme. Ha recibido honorarios por conferencias de algunas de las industrias mencionadas.
Dr. Dubrovsky: Ha recibido honorarios por asesoramiento científico de los laboratorios PTC, Sarepta, Biogen, Sanofi Genzyme, Takeda Avexis, Novartis, Raffo, Roche. Ha recibido becas para investigación por parte de Genzyme-Sanofi, PTC, Novartis, Sarepta, Biogen. Ha recibido honorarios por conferencias de algunas de las industrias antes mencionadas.

