










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957
Acta bioquím. clín. latinoam. vol.47 no.3 La Plata set. 2013
TOXICOLOGIA
Dependencia de la dosis en los mecanismos de toxicidad y la evaluación de riesgo en toxicología
Dose-dependent transitions in mechanisms of toxicity and risk assessment in toxicology
Dependencia da dosagem nos mecanismos de toxicidade e avaliagáo de risco em toxicologia
Gerardo Daniel Castro1
1. Doctor en Ciencias Químicas
Instituto de Investigación e Ingeniería Ambiental. Universidad Nacional de General San Martín (UNSAM). 25 de Mayo y Matías de Irigoyen, 1650 SAN MARTÍN, Buenos Aires, Argentina.
Centro de Investigaciones Toxicológicas (CEITOX-UNIDEF, MINDEF-CONICET). Juan B. de La Salle 4397, 1603 Villa Martelli, Buenos Aires, Argentina.
Abreviaturas
ADI ("acceptable daily intake"): Es una medida de la cantidad de una sustancia (en general se lo aplica al caso de un aditivo alimentario, aunque también se lo menciona en el caso de fármacos veterinarios o pesticidas) en un alimento o en agua de bebida que puede ingerirse sobre una base diaria por toda la vida sin que ocurra un riesgo apreciable para la salud. Se la expresa usualmente en miligramos por kilo de peso corporal por día.
BMDL ("benchmark dose lower confidence limit"): Este concepto provee una alternativa más cuantitativa en el primer paso de la evaluación dosis-respuesta que el proceso actual NOAEL/LOAEL para efectos no-cáncer (U.S. EPA, 1996). En tanto la EPA está cambiando sus criterios hacia la armonización de los criterios de evaluación para efectos carcinogénicos y no-cáncer, se considera mejor considerar el modo de acción y si los efectos tienen un comportamiento linear o no a dosis bajas.
ED10: Dosis efectiva 10. Se dice de aquella dosis que produce el efecto en el diez por ciento de los individuos bajo estudio.
Hormesis.- Fenómeno de respuesta a la dosis que se caracteriza por una estimulación por dosis bajas y una inhibición para dosis altas, lo que resulta en una curva de respuesta en forma de J o de U invertida. Un tóxico que produzca el efecto de hormesis tiene entonces a bajas dosis el efecto contrario al que tiene en dosis más altas. El principio de funcionamiento de la hormesis no siempre está claro. A menudo se consideran dos efectos completamente contrarios que funcionan en paralelo: un efecto positivo que ya se presenta en dosis muy pequeñas, y un efecto negativo que sólo aparece con las dosis más grandes.
LOAEL (lowest observed adverse effect level): Es la concentración o cantidad más pequeña de una sustancia que causa una alteración observable y distinguible de un control apropiado.
NOAEL (no observed adverse effect level): Es la dosis más elevada de una sustancia que no ha mostrado en las pruebas tener efectos perjudiciales para la salud en personas o animales.
PBPK (physiologically based pharmacokinetic modeling): El modelado farmacocinético basado fisiológicamente es una metodología de modelamiento matemático de la far-macocinética para predecir la absorción, distribución, metabolismo y excreción de un compuesto en humanos y otras especies animales. El modelado PBPK se usa en investigación y desarrollo farmacéutico, así como para la evaluación de riesgo en la salud.
RfD (reference dose): Es la dosis oral máxima aceptable de una sustancia tóxica, según la Agencia de Protección Ambiental de EE.UU. (U.S. EPA). Se la determina frecuentemente para los pesticidas y se la define como una estimación, con una incertidum-bre que puede abarcar un orden de magnitud, de una exposición diaria oral de la población humana (incluyendo subgrupos sensibles) que es probable que no tenga un riesgo apreciable de efectos dañinos durante toda la vida.
Resumen
En la Toxicología se presentan desafíos derivados de la complejidad creciente de los escenarios ambientales que condicionan a muchas enfermedades humanas. Cuando se analiza la curva dosis-respuesta tóxica, frecuentemente se hace evidente que pueden existir varios procesos subyacentes para la acción de cualquier xenobiótico sobre un organismo vivo. Es muy probable que los pasos críticos, limitantes en cualquier mecanismo de toxicidad, puedan verse sobrepasados con exposiciones más grandes, señalando esto la emergencia de modos nuevos de injuria tisular a dosis más altas. Por lo tanto, pueden ocurrir transiciones dependientes de la dosis en el mecanismo principal de toxicidad, que tendrían un impacto significativo en la interpretación de la colección de los datos de referencia para la evaluación del riesgo. Una explicación para las relaciones lineales dosis-respuesta en serie es la transición dosis-dependiente entre una secuencia de pasos encadenados mecanística-mente, pasos limitantes de la velocidad saturables en el proceso total, que llevan desde la exposición a la expresión de uno o más modos para la respuesta tóxica. El análisis de estos fenómenos tiene una relevancia muy grande en términos prácticos, esto es, para las consecuencias sobre la regulación del uso y exposición a sustancias peligrosas en distintos ámbitos humanos.
Palabras clave: Evaluación de riesgo; Mecanismos de toxicidad; Biomon/toreo; Dosis-respuesta
Summary
In Toxicology, many challenges arise from the increasing complexity of environmental scenarios conditioning human diseases. When analyzing the toxic dose response, it often becomes apparent that there may be several underlying processes for any xenobiotic action on a living organism. It is likely that the critical steps, limiting in any mechanism of toxicity, may be overwhelmed with increased exposure, indicating the emergence of new forms of tissue injury at higher doses. Therefore, transitions might occur in a dose-dependent manner in the main mechanism for toxicity, having a significant impact on the performance of the collection of baseline data for risk assessment. One explanation for the linear dose-response relationships in series is dose-dependent transition from a sequence of steps chained mechanistically, saturable rate limiting steps in the process, leading from exposition to the expression of one or more forms of toxic response. The analysis of these phenomena has a great relevance in practical terms, that is, to the effects on the regulation of the use and exposure to hazardous substances in different human situations.
Keywords: Risk assessment; Mechanisms of toxicity; Biomonitoring; Dose response
Resumo
Em Toxicologia apresentam-se desafios decorrentes da complexidade crescente dos cenários ambientais que condicionam muitas doengas humanas. Ao analisar a curva dosagem-resposta tóxica frequentemente se torna evidente que podem existir vários processos subjacentes para a agáo de qualquer xenobiótico num organismo vivo. É muito provável que os passos essenciais, limitantes em qualquer mecanismo de toxicidade, possam ser superados com exposigoes maiores, indicando o aparecimento de modos novos de lesáo tissular com doses mais altas. Portanto podem acontecer transigoes que dependam da dosagem no mecanismo principal de toxicidade, que teriam um impacto significativo na interpretagáo da coleta de dados de referencia para a avaliagáo do risco. Uma explicagáo para as relagoes lineares dose-resposta em série é a transigáo dose-dependente entre uma sequencia de passos encadeados mecanicamente, passos limitantes da velocidade saturáveis no processo total, que levam da exposigáo a expressáo de um ou mais modos para a resposta tóxica. A análise desses fenómenos tem grande relevancia em termos práticos, isto é, para as consequencias na regulagáo do uso e exposigáo a substancias perigosas em diferentes ámbitos humanos.
Palavras-chave: Avaliacao de risco; Mecanismos de toxicidade; Biomonitoramento; Dosagem-resposta
Introducción
En la Toxicología de hoy se presentan desafíos derivados de la complejidad creciente de los escenarios ambientales que condicionan a muchas enfermedades humanas. El cáncer es una de las más relevantes pero no debe olvidarse a varias patologías del sistema nervioso e inmunológico, del sistema reproductor en ambos sexos, la teratogénesis, todos problemas donde se sospecha con buena razón la culpabilidad de la exposición creciente de las poblaciones a mezclas de sustancias químicas. La epidemiología clásica no puede enfrentar el análisis del riesgo implícito en estas situaciones tan complejas porque sus herramientas de diagnóstico no permiten discriminar fácilmente las causas múltiples ni prever las consecuencias de la exposición a sustancias químicas por distintas vías, y a dosis que seguramente no producirían efectos inmediatos o visibles. Por otra parte, cuando se necesita adoptar una regulación deben establecerse primero niveles de exposición seguros, concentraciones límite aceptables, etc. Ese aspecto "cuantitativo" de la evaluación de riesgo toxicológico requiere del uso de herramientas que permitan medir, ya no la presencia del tóxico en el ambiente, sino al tóxico en alguna forma en el organismo expuesto (toxicocinética) o a las consecuencias deletéreas (toxicodinamia). La epidemiología molecular contribuye con la salud pública mejorando la evaluación de la exposición (lo cual tiene consecuencias directas sobre la prevención primaria), identificando criterios de valoración tempranos de enfermedad (los que pueden utilizarse con propósitos de rastreo o ensayos clínicos), e identificando individuos que presentan un riesgo aumentado para la enfermedad, y entonces puede ayudar a mejorar la determinación del riesgo proveyendo mejores relaciones exposición respuesta e información sobre susceptibilidad. Además, incorporando biomarcadores que puedan identificar exposiciones, alteraciones y susceptibilidad a nivel celular, la epidemiología molecular permite una comprensión de los mecanismos implícitos en el proceso de la enfermedad. Esta disciplina ha provisto los argumentos racionales para establecer niveles de riesgo razonables frente a exposiciones ambientales, ocupacionales, alimentarias, etc. (1).
En la medida que se acumula la experiencia con la comprensión de los mecanismos de toxicidad, se hace evidente que pueden existir varios procesos subyacentes para la acción de cualquier xenobiótico sobre un organismo vivo cuando se analiza la curva dosis-respuesta tóxica. Es muy probable que los pasos críticos, limitantes de un mecanismo de toxicidad dado, puedan verse sobrepasados con exposiciones incrementadas, señalando esto la emergencia de modos nuevos de daño tisular a dosis más altas. No debe resultar extraño entonces postular que pueden ocurrir transiciones dependientes de la dosis en el mecanismo principal de toxicidad, que tendrían un impacto significativo en la interpretación de la colección de los datos de referencia para la evaluación del riesgo. Una explicación para las relaciones lineales dosis-respuesta en serie es la transición dosis-dependiente entre una secuencia de pasos encadenados mecanísticamente, pasos limitantes de la velocidad saturables en el proceso total, que llevan desde la exposición a la expresión de uno o más modos para la respuesta tóxica. El análisis de estos fenómenos tiene una relevancia muy grande en términos prácticos, esto es, para las consecuencias sobre la regulación del uso y exposición a sustancias peligrosas en distintos ámbitos humanos (2) (3). Lo que se vuelve particularmente desafiante para la evaluación del riesgo es si dicha transición en la linealidad ocurre entre la dosis experimental a partir de la cual el riesgo se extrapola, y la dosis a la cual los humanos se verían expuestos. En estas situaciones, es imperativo que los protocolos que se desarrollen para ayudar en la estimación del riesgo incluyan consideraciones acerca de la ocurrencia de dichas transiciones, esperada en los pasos limitantes clave en el mecanismo global de toxicidad del compuesto en cuestión. Nuevamente, serán los biomarcadores la herramienta más idónea para identificar y caracterizar estos fenómenos.
¿QUÉ SE ENTIENDE POR TRANSICIONES?
Para los propósitos de este análisis, puede definirse una "transición" como un cambio o variación con dosis incrementadas, en los factores cinéticos o dinámicos clave subyacentes, que influyen el mecanismo responsable para la tasa de respuesta en función de la dosis. Una transición ocurre usualmente dentro de un rango de dosis y refleja un continuo de cambio, más que un simple punto de partida. La demostración y la caracterización de una transición dependiente de la dosis deberían influir en la evaluación de los datos, tanto por encima como por debajo de la transición (2).
El concepto es que cada proceso inducible o saturable que ocurre como parte de un mecanismo de la acción tóxica y la respuesta biológica total, representa un punto de partida potencial para abandonar la linealidad en la relación dosis-respuesta. Los ejemplos cinéticos de procesos saturables y/o inducibles incluyen absorción, distribución (unión con las proteínas, transporte activo), eliminación y transformación química (activación y detoxificación). Los ejemplos dinámicos incluyen interacciones con el receptor (constantes de afinidad, un número finito de receptores, tipos de receptor múltiples), reparación/reversión del daño (reparación del ADN, reactivación del receptor, síntesis proteica, reemplazo celular) y homeostasis alterada (inducción, cambio metabólico, proliferación celular y apoptosis). En el curso de esta revisión se analizan varios ejemplos, en la forma de análisis de casos, de cómo ocurren las transiciones en los mecanismos dependientes de la dosis. Estos ejemplos representan algunos de los distintos mecanismos de las transiciones dosis-dependientes. Sin embargo, los estudios de estos casos no ejemplifican todos los mecanismos posibles. Los estudios de casos proveen una oportunidad para describir el impacto potencial de este fenómeno en el proceso de evaluación del riesgo, al proveer aproximaciones a la integración de estas curvas dosis-respuesta no lineales dentro del proceso de evaluación del riesgo. La razón para esto es que los aspectos concernientes al diseño experimental apropiado y a los requerimientos de datos están delineados para conjuntos de datos que exhiben transiciones dependientes de la dosis en los mecanismos de toxicidad.
A partir de los datos científicos se espera construir un entendimiento de este fenómeno y buscar un campo común entre los expertos de la industria, académicos y del Estado acerca de los principios y los métodos para incorporar estos fenómenos en las decisiones de evaluación del riesgo.
RELEVANCIA DEL FENÓMENO EN LA EVALUACIÓN DEL RIESGO TOXICOLÓGICO
Los efectos de las dosis altas empleadas en los estudios experimentales con animales son a menudo difíciles de interpretar y pueden no reflejar la toxicidad real a niveles de exposición humana relevantes. En particular, esto puede ser cierto si las transiciones dependientes de la dosis ocurren en el principal mecanismo responsable de la toxicidad. Para evaluar este proceso críticamente, se necesitan describir y evaluar ejemplos de sustancias y clases químicas para los cuales haya evidencia firme que indique que a dosis diferentes existen mecanismos diferentes. La atención debería enfocarse en el concepto de los "gatillos" (disparadores) biológicos para eventos o pasos clave, moduladores de elementos críticos en un modo dado de acción. Este concepto tiene una importancia directa para descifrar la relevancia de estudios con dosis "más altas" en un ensayo tradicional de toxicidad para la probabilidad de eventos adversos bajo los escenarios reales de exposición humana.
EJEMPLOS DE TRANSICIONES DE MECANISMOS SATURABLES O INDUCIBLES
El proceso que va desde la exposición a una sustancia química hasta la expresión de una respuesta biológica consiste en una serie de pasos interrelacionados aunque cinéticamente independientes, cada uno de los cuales tiene su propio conjunto de características de dependencia con la dosis (o concentración). Los ejemplos de procesos relacionados secuencialmente incluyen la absorción del agente a través de la superficie expuesta, su reparto entre moléculas transportadoras específicas dentro del espacio vascular, la distribución perfusión-dependiente a todos los órganos, el pasaje del agente desde el espacio vascular hasta el tejido, la distribución tejido-específica incluyendo la asociación con elementos de unión específicos dentro de cada tejido, la bio-transformación del compuesto a través de uno o más procesos enzimáticos, que ocurren tanto en paralelo (compitiendo) como en serie (cuando el producto de una reacción es el sustrato para la siguiente), el movimiento de cualquier metabolito tanto dentro como desde el tejido, la redistribución del xenobiótico o de sus metabolitos dentro y a través de la circulación, y finalmente, la eliminación del compuesto original o de sus metabolitos desde el organismo expuesto. Los procesos saturables también ocurren sobre una base dinámica e incluyen la existencia de un número finito de moléculas receptoras o una capacidad limitada para restaurar la molécula blanco a su forma nativa (disociación del receptor, reactivación, reparación o regeneración).
Aunque hay varios ejemplos de procesos no saturables pasivos individuales que ocurren en el transcurso que lleva definir la relación dosis-respuesta para cualquier sustancia dada, es extremadamente raro que cada paso dentro del camino sea de orden cero respecto a la dosis. Consecuentemente, no es tanto que las curvas dosis-respuesta se desvían desde una única y simple relación lineal logarítmica de primer orden por encima del rango total de concentraciones de exposición. Es más típico que tales relaciones consistan en múltiples relaciones de primer orden a medida que uno explora el rango total de dosis (Fig. 1).

Figura 1. Representación conceptual de los factores a considerar en relación con transiciones dependientes de la dosis en determinantes de toxicidad.
La Tabla I muestra varios pasos dentro de la cinética de disposición y de la expresión dinámica de la toxicidad para varios xenobióticos junto con ejemplos de sustancias de las que se sabe que exhiben transiciones dosis-dependientes para esos procesos.
Tabla I. Algunos ejemplos de procesos y sustancias químicas que están sujetos a transiciones dependientes de la dosis en la disposición cinética y la expresión dinámica.

No linealidades con dosis altas y linealidad con dosis bajas en la cinética de los carcinógenos genotóxicos
En carcinogénesis, es común en la evaluación del riesgo carcinogénico expresar la dosis en términos de tasa diaria promedio a lo largo de la vida. En los hechos, ni las exposiciones de los animales experimentales ni las exposiciones humanas en la vida real, siguen una tasa constante por toda una vida. Puede por lo tanto haber cambios en la reactividad del ADN, la mutación somática y el riesgo de cáncer resultante, que pueden esperarse de estas fluctuaciones. La discusión presente se limitará a los procesos que contribuyen a la carcinogénesis hasta la producción de las transiciones mutagénicas. No se abordará aquí el tema de las diferencias sustanciales en el riesgo carcinogénico que pueden producirse por la inducción de transiciones tempranas o tardías en el proceso carcinogénico a edades diferentes.
¿Por qué debería esperarse una linealidad básica en las reacciones con el ADN a dosis bajas? Respuesta: la química...
Imagínese que por algún proceso, una pequeña cantidad de sustancia X capaz de reaccionar con el ADN,
pudiera entregarse directa y continuamente dentro del núcleo de la célula. Después de un corto período de equilibrio, el ingreso constante de X dentro del núcleo podría balancearse por la reacción de X con el ADN y otros nucleófilos y la salida de X fuera del núcleo, de tal forma que la concentración de X debería alcanzar un estado estacionario.
Bajo estas circunstancias, la reacción entre X y el ADN estará regida por una cinética de reacción bi-molecular ordinaria. Las moléculas de los reactivos colisionarán al azar unas contra otras, y el número de dichas colisiones dependerá directamente de [X] y [ADN] (o de los sitios del ADN reactivos relevantes). La tasa absoluta de la reacción dependerá de la fracción de colisiones que ocurren en los ángulos correctos y que tienen suficiente energía (en otras palabras, la "energía de activación") para sobrepasar las fuerzas repulsivas que intentan mantener apartadas a las moléculas. Mientras [X] no sea lo suficientemente alta para consumir apreciablemente el número de los sitios más reactivos del ADN, entonces la tasa de la reacción total con todos los sitios del ADN debe ser una función lineal simple de [X].
El tema no es tan simple, de todos modos. A pesar de lo que se esperaría sobre la linealidad en el proceso de reacción con el ADN, hay varios otros pasos a lo largo del camino causal hacia la carcinogénesis, tanto antes como después de la reacción con el ADN, que puede esperarse que produzcan no-linealidades a dosis altas. Esto es lo que se discutirá en las secciones siguientes.
Cambios en los procesos de captación del tóxico a dosis diferentes
Transporte pasivo simple. Los procesos de transporte pasivo deben ser generalmente funciones lineales de la tasa de la dosis, a dosis razonablemente bajas. Este caso es bastante similar a la reacción del ADN descrita más arriba. Representando a una membrana como una barrera con un número fijo de poros, puede esperarse entonces que la tasa a la cual las moléculas pasan desde un lado al otro de la barrera sea directamente proporcional al número de "colisiones" entre una molécula de la sustancia difundida y una de las "puertas" en la barrera. Mientras que la concentración de la sustancia sea lo suficientemente baja para que las propiedades principales de la membrana permanezcan sin cambio, y el número de poros libres disponible en cualquier momento no esté disminuido apreciablemente, entonces no debería haber efecto debido a la tasa de la dosis.
Transporte facilitado de sustancias dentro del organismo. Cuando los procesos de transporte son facilitados por un número finito de macromoléculas transportadoras, es esperable que ocurra (a) un aumento del transporte a niveles bajos de la concentración externa, relativo a lo que podría ocurrir en ausencia de un sistema de facilitación, y (b), una saturación de la porción de ese transporte facilitado a rangos de dosis relativamente altos. Si el transporte es facilitado por un sistema saturable simple (como por ejemplo, la absorción de un ion metálico pesado desde el intestino a través de un sistema cuya función normal es el transporte de calcio), entonces la dosis entregada mostrará un patrón similar al de la saturación de una enzima activadora, o sea, la tasa de provisión del tóxico al sistema se aproximará a una asíntota (Vmáx):
dS/dt = (Vmáx [S]externa) / (K + [S]externa)
donde S es el sustrato. Sin embargo, debería señalarse que estos tipos de procesos deben todavía proceder linealmente al límite de dosis bajas (con una tasa constante de Vmáx/K), donde el número de sitios de unión/ transportadores ocupados es pequeño en relación con el total de sitios disponibles.
El transporte también puede afectarse a dosis altas por las respuestas fisiológicas, tales como la irritación, similar a las tasas de respiración decrecientes en animales observadas en respuesta a algunos tóxicos en el aire. Por esta razón, para el óxido de etileno en la experimentación animal se encontró necesario, para modelar estos efectos a dosis altas, acomodar los datos disponibles acerca del ingreso y la formación de productos. Estos efectos tienden a desaparecer con dosis bajas (4).
Cambios en la excreción dependientes de la dosis
La excreción comprende la eliminación física de sustancias desde el cuerpo, ya sea vía los riñones en la orina, vía el camino bilis-intestino-heces, o menos comúnmente, vía el pelo, la transpiración o la saliva. En gran medida, la mayoría de las no linealidades de este tipo se relacionan con la excreción urinaria. Se conocen dos tipos de procesos de transporte activo saturables y que juegan roles importantes en la eliminación urinaria de algunos compuestos, la secreción tubular y la reabsorción tubular. La saturación de la secreción tubular tiende a disminuir el clearance renal a dosis altas, y por lo tanto produciría concentraciones corporales internas más grandes a tasas mayores de dosis externas, mientras que la saturación de los procesos de reabsorción tubular provocaría el efecto opuesto. Debido a que la secreción tubular tiene el efecto de hacer que una exposición de corto tiempo y dosis alta sea relativamente más riesgosa que la misma dosis entregada a lo largo de un período más largo, es particularmente importante para un sistema de screening reconocer situaciones donde puede ocurrir este tipo de comportamiento de saturación.
Quizás, la mejor aproximación simple para el reconocimiento preliminar de situaciones donde la saturación de la secreción tubular pueda ser importante es administrar una dosis alta en bolo del compuesto y seguir la concentración sanguínea y la excreción urinaria en el tiempo. Después de lograr un equilibrio inicial entre la sangre y los tejidos corporales, la saturación de la eliminación estará señalada por un aumento en la pendiente de la caída del logaritmo de la concentración sanguínea con el tiempo, esto es, una curva con dirección hacia abajo en un diagrama del logaritmo de la concentración sanguínea vs. tiempo. Debido a que dicho cambio en la tasa de declinación puede ser producido también por la saturación de un paso de eliminación metabólica, la confirmación de la causa de la declinación de la saturación de un paso involucrado en la excreción urinaria sólo puede hacerse si los datos de la excreción urinaria indican que este camino da cuenta de una fracción relativamente alta de la eliminación global a todas las dosis relevantes.
Otros fenómenos de saturación producen los resultados opuestos para el clearance de dosis altas, con un incremento del riesgo para niveles sanguíneos elevados, tendiendo a reducir la dosis interna por tiempo por unidad de dosis a tasas altas de dosis. Estos fenómenos incluyen tanto la reabsorción tubular y, completamente por fuera del riñón, la saturación de la unión a los glóbulos rojos y a las macromoléculas en sangre. Por ejemplo, se ha visto que a concentraciones sanguíneas altas de plomo hay un cociente más alto de plomo plasmático sobre el de plomo en los glóbulos rojos (5). Esto está relacionado probablemente con el aumento en la tasa de clearance renal de plomo a niveles sanguíneos altos de plomo, fenómeno reportado en algunas investigaciones (6).
Cambios dependientes de la dosis en el metabolismo
Cinética enzimática de Michaelis-Menten. Implicancias teóricas. Las enzimas que son responsables de activar o detoxificar los xenobióticos tienen capacidades finitas. A medida que se incrementan las concentraciones de un sustrato, y las enzimas metabólicas clave comienzan su aproximación asintótica a la saturación, progresivamente porciones mayores de la sustancia administrada quedan sin metabolizar o disponibles para ser procesadas por rutas secundarias, con una aproximación más lenta a la saturación o sin comportamiento para nada saturante (por ejemplo, la exhalación de una sustancia volátil).
La ecuación básica de saturación de Michaelis-Menten no es simplemente una fórmula empírica, sino una teoría bien fundamentada en consideraciones mecanísticas de asociación del receptor y cinética de disociación, con amplia aplicabilidad. Existen casos específicos en los cuales pueden predecirse las implicancias de la cinética de Michaelis-Menten, a saber:
1. En el caso que una enzima activa el sustrato a un intermediario tóxico.
1A. A tasas de dosis "bajas", alguna fracción constante del sustrato se convertirá en el tóxico proximal (esto es, la forma activa), con la proporción exacta dependiendo del cociente de la velocidad constante para la reacción activadora (V /K ) sobre la velocidad constante de la eliminación total del sustrato del sistema, incluyendo exhalación, excreción urinaria, otras enzimas, etc. Este punto puede entenderse tratando específicamente el caso donde una enzima activadora saturable está compitiendo con una enzima detoxificante saturable.
La velocidad de la reacción activadora (que produce el metabolito activo, A) es:
dA/dt = (Vmáx[S]) / (Km + [S])
De manera similar, la velocidad de la reacción de de-toxificación que compite (la que produce el metabolito inactivo, I) es:
dI/dt = (Vmáx’ [S]) / (Km’ + [S])
Las [S] en el denominador pueden despreciarse a dosis bajas cuando se tornan pequeñas en relación con Km. A dosis bajas, el cociente del sustrato [S] que va por los caminos metabólicos de la molécula activa o la molécula inactiva se define simplemente dividiendo las dos ecuaciones:
dA/dt / dI/dt = Vmáx[S]/Km / Vmáx’[S]/K’m
Debido a la supresión de las [S], queda un cociente de cuatro constantes. Por debajo de la región de dosis donde hay saturación apreciable, una fracción constante del sustrato debe ser tomada por cada camino, independientemente de la dosis. No hay efectos de la tasa de la dosis para la formación del tóxico proximal en esta región de dosis bajas.
1B. Por otro lado, a tasas de dosis "altas", habrá alguna velocidad absoluta máxima de generación de la molécula activa, como se sugirió para el caso del cloruro de vinilo. Este concepto se ha usado para explicar el hecho de que aunque los experimentos iniciales con el cloruro de vinilo produjeron relaciones dosis-respuesta esencialmente lineales para tumores por debajo de las 500 ppm, por encima de este nivel la respuesta tumoral fue esencialmente plana (hasta las 6000 a 10000 ppm), y se observó poco riesgo tumoral adicional (7) (8). Si, como usualmente es el caso, sólo hay disponibles dos niveles de dosis altas, y si se han desarrollado los procedimientos estándar recomendados para la interpolación lineal del riesgo de cáncer desde los niveles de dosis de 6000 a 10000 ppm, el riesgo real para las ratas a la dosis "baja" (250 ppm) podría haber sido subestimado, y el incremento del riesgo (pendiente de la curva dosis-respuesta) también habría sido subestimado por varios órdenes (Fig. 2). La misma proyección podría haber llevado a una sobreestimación de la dosis asociada con un nivel de riesgo particular (por ejemplo, una ED10).

Figura 2. Subestimaciones y sobrestimaciones del riesgo de cáncer para la exposición al cloruro de vinilo. Referencias: (7) (8).
En otro ejemplo relevante, se ha observado una saturación aparente similar del metabolismo activante del carcinógeno NNK (4-(N-metil-N-nitrosamino)-1-(3-piridil)-1-butanona), una nitrosamina relacionada con el hábito de fumar tabaco. En la mucosa nasal de la rata, se encontró que NNK produce compuestos de O6-metilguanina con una eficiencia ocho veces mayor por unidad de dosis a dosis (intraperitoneales) bajas (0,3-1 mg/kg) que a 100 mg/kg. Se ha sugerido que esto puede atribuirse a la saturación de una enzima local que produce el agente metilante activo, ya que no se ve otra dependencia de la dosis tan dramática similar en la mucosa olfatoria vecina (9).
2. Por otra parte, las siguientes consecuencias ocurren si una enzima saturable es la responsable por la detoxificación.
2A. A tasas de dosis "altas", el compuesto original o el metabolito biológicamente reactivo tendrán una tasa de eliminación más lenta del cuerpo por unidad de concentración, y por lo tanto, más oportunidad de reaccionar con las moléculas blanco, que con tasas de dosis menores. Si las rutas alternativas de pérdida del sustrato desde el sistema son procesos de primer orden no saturables (por ejemplo, exhalación) operando a una tasa combinada, k', entonces el cociente de la dosis entregada sobre la dosis externa en el límite de los valores de dosis altas se incrementará por encima del cociente correspondiente a valores de dosis bajas por el factor siguiente:
(k’ + Vmáx/Km) / k’
En contraste, si no hay o hay solamente rutas de eliminación del sustrato muy menores no saturables, entonces la declinación en la concentración del sustrato después de una gran dosis inicial será esencialmente lineal con el tiempo (a la velocidad V ). Si la concentración en el cuerpo al comienzo es [S]0, entonces al menos inicialmente (hasta que la alta concentración inicial disminuya lo suficiente para que la enzima no esté saturada de modo apreciable), se verificará que:
[S]t = [S]0 - Vmáx t
Para una primera aproximación, entonces, un diagrama de concentración vs. tiempo será lineal, intersecando la ordenada en [S]0, y la abscisa en [S]0/Vmáx. Debido a que la intersección con la abscisa es directamente proporcional a la altura [S]0, se desprende que el área triangular por debajo de la línea es proporcional al cuadrado de la altura, esto es, el cuadrado de la dosis inicial:
Aductos de ADN formados = K [S]0 2/ 2 Vmáx
Por lo tanto, las dosis agudas altas pueden causar una respuesta [dosis]2, comparada con la respuesta lineal observada a dosis bajas, debajo de lo necesario para saturar las enzimas detoxificantes o reparadoras clave.
Cambios dependientes de la dosis en la eficiencia de la reparación, la muerte celular y la tasa de proliferación celular
Reparación del ADN. Si todo lo demás es igual, la saturación de los sistemas de reparación a tasas altas de generación de una lesión tiende a llevar a una persistencia mayor de las lesiones y a una mayor oportunidad de las lesiones para estar presentes en el momento de la copia del ADN, cuando los cambios permanentes o los reordenamientos de la información codificada en su estructura pueden ser fijados en el genoma. Esto podría producir una curva de mutaciones fijas en relación con la tasa de dosis carcinogénica con dirección hacia arriba. Inherentemente, sin embargo, los sistemas de reparación del ADN no pueden alcanzar la reparación completa incluso a muy bajas tasas de daño del ADN. Si (a), hay un número finito de moléculas enzimáticas capaces de reconocer un tipo particular de lesión del ADN e iniciar el proceso de reparación y (b), hay sólo un lapso finito entre la generación de la lesión y la potencial concreción del proceso mutagénico a través de la terminación del proceso de la división celular, entonces en general alguna fracción finita de las lesiones reparables a dosis bajas escapará a la reparación.
Asumiendo que el reconocimiento y la reparación de la lesión están gobernados por los mismos principios básicos que otras reacciones enzimáticas, si hubo una simple "explosión" en la generación de lesiones en el tiempo t = 0, la tasa de reparación de lesión (-d[L]0/dt) podría estar dada por:
d[L]/dt = Vmáx [L] / (Km + [L]')
lo cual, por supuesto, producirá las mismas implicancias para las proyecciones de dosis altas a dosis bajas, como se discutió anteriormente para las reacciones de detoxificación y para la secreción tubular activa. A dosis bajas, las lesiones generadas inicialmente disminuirán exponencialmente con una tasa constante igual a V / máx Km. Si hay un dopaje continuo de nivel bajo, entonces la tasa de generación de lesiones (k-[S]interna) llegará a un equilibrio con la tasa de reparación de lesiones. La concentración de lesiones en el equilibrio puede representarse como:
[L]eq = k [S]interna Km/Vmáx
La tasa de generación de mutaciones somáticas permanentes a dosis bajas debe ser simplemente proporcional a esta cantidad multiplicado por la tasa de repli-cación de las células relevantes. Para dosis altas únicas, la generación masiva de lesiones excede por lejos las dosis requeridas para saturar las enzimas relevantes de reparación. Con el mismo razonamiento que se aplica para la detoxificación, puede esperarse tanto como una dependencia cuadrática [lesión]20 con la concentración por el tiempo para la producción de lesiones en el ADN, exacerbado esto por el aumento que puede haber en la replicación celular a dosis altas (algo que se analizará más adelante).
En la literatura se han reportado observaciones que sugieren la inducción del sistema de reparación del ADN en las células linfoblásticas humanas expuestas in vitro a la aflatoxina B1 (10). En este sistema, se encontró que la pérdida de aductos de N7-guanina después de la exposición era bifásica, mostrando inicialmente una vida media de tres a cuatro horas, seguido por una tasa de pérdida más lenta con una vida media de más o menos diez horas. A tres dosis diferentes, dando tres niveles iniciales diferentes de aductos de N7-guanina, el cambio desde la fase de pérdida más rápida hasta la más lenta (correspondiente presumiblemente a la reversión de la concentración enzimática hasta niveles no inducidos) ocurría cuando quedaban más o menos 1000 aductos totales por célula. En general, la inducción de un sistema de reparación de bajo error relativo, como en este caso, puede esperarse que lleve a una saturación de las transiciones mutagénicas como una función de la dosis. Por el contrario, si el sistema de reparación inducible es más propenso al error que los procesos constitutivos para remover los aductos, la no linealidad irá en la dirección de producir una curva de dirección ascendente de transiciones mutagénicas en relación con la dosis. De una manera similar, la inducción de enzimas metabólicas detoxificantes o activantes podría tener consecuencias análogas para las relaciones dosis-respuesta.
Una interpretación en términos de una reparación inducible de ninguna manera carece de incertidumbre. De hecho, los fenómenos de cinéticas bifásicas se han observado repetidamente en años recientes. Estas observaciones se interpretaron como que las diferencias en las tasas de reparación del ADN para aductos similares pueden ser diferentes para aductos localizados en una cromatina relativamente condensada versus una no condensada, en células en diferentes fases del ciclo celular, en células que están ciclando versus las que no lo están, o en otros "compartimientos" del ADN.
Los casos de una reparación de ADN inducible o de actividades enzimáticas de detoxificación inducibles tienen otra consecuencia posible que merece mencionarse. Es teóricamente factible que en algunos casos donde hay un background de daño en el ADN apreciable por agentes distintos del tóxico en cuestión, por encima de cierto rango de dosis, la inducción de las actividades enzimáticas de defensa puede causar reducciones apreciables en los procesos carcinogénicos de fondo. En algunos casos, por encima de cierto rango de dosis, es incluso posible que estas reducciones en la carcinogénesis "de fondo" sean cuantitativamente mayores que los aumentos en los procesos de cáncer que se esperarían de la acción directa del carcinógeno inductor, causando un aparente efecto de "hormesis". Las observaciones de hormesis, incluyendo algunos ejemplos aparentes para puntos finales carcinogénicos, han sido sujeto de discusión muy importante, por razones obvias. En casos donde la inducción de enzimas de detoxificación o reparación ocurre en los bioensayos experimentales de cáncer, y donde puede mostrarse que estos producen un efecto discernible en las frecuencias background del tumor, entonces el tratamiento adecuado del modelo podría ser análogo al tratamiento de no linealidades cinéticas para dosis altas. Esto significa que estas influencias extrañas sobre las frecuencias tumo-rales observadas deberían (idealmente) ser separadas estadísticamente de las observaciones dosis-respuesta totales antes de evaluar el efecto carcinogénico directo del compuesto inducido con los modelos de etapas múltiples.
Muerte celular y aumento de la replicación celular. El incremento de la replicación celular bien puede ser una de las fuentes más comunes para las no linealidades para las dosis altas. Los protocolos actuales para realizar bioen-sayos de carcinogénesis, de dos a cuatro veces las "dosis máximas toleradas" estimadas, algunas veces llevan a probar dosis que inducen una muerte celular apreciable e incrementos en la división celular en los tejidos blanco de la acción carcinogénica. La curva dosis-respuesta muy empinada de la carcinogénesis por formaldehído es un caso para ilustrar este punto. En experimentos relativamente prolongados con marcación de timidina, conducidos por un período de seis días con la ayuda de una minibomba osmótica implantada, Swenberg et al. (11) encontraron que mientras sólo el 0,19% de las células epiteliales respiratorias en las ratas control habían sintetizado ADN, el 13,5% de células similares se habían marcado en las ratas expuestas por cinco días a 15 ppm de formaldehído. Este incremento de 71 veces en la tasa de replicación celular podría estar asociado teóricamente con un incremento similar de la tasa de fijación de lesiones del ADN inducidas por el formaldehído a dosis altas y tener un efecto marcado en la interpolación de la incidencia de formación tumoral entre exposiciones altas y bajas. Debe notarse, sin embargo, que esto es estrictamente una interpolación, no una extrapolación, debido a que los datos requeridos del punto cero exceden los tumores sobre un background a dosis cero.
Usando una aproximación más convencional para el experimento de marcación con timidina radiactiva (dando la marcación dos horas antes de la eutanasia), se generaron datos sobre la dependencia con el tiempo para la respuesta de proliferación celular a exposiciones diarias durante 5 días por 6 horas a 200 ppm de bromuro de metilo (12). En este caso, la respuesta hizo un pico en el día 3 antes que las exposiciones se completaran (debido al reemplazo del epitelio respiratorio destruido con células escamosas). Es también de interés que el epitelio no retornó a los niveles basales de re-plicación celular hasta más de un mes después del período de exposición, proveyendo un tiempo extendido para posibles interacciones entre diferentes episodios de corta duración y alta exposición. En el caso de radiaciones ionizantes, las diferencias en las tasas de proliferación celular probablemente hayan contribuido a un riesgo aparente de carcinogénesis 10 veces más alto por unidad de dosis de rayos X para fetos humanos expuestos en el primer trimestre, en comparación a fetos expuestos en el segundo y el tercer trimestre (13).
Resumiendo, hay razones teóricas sólidas para explicar porqué todos los procesos cinéticos deben "ir lineales" en el límite de las dosis bajas. A tasas de dosis bajas, los procesos de transporte y de transformación que llevan al daño y la reparación del ADN dependen directamente del número de colisiones entre las moléculas de un tóxico "ingresado" (o aducto de ADN o intermediario activado) y un reactivo celular residente (o un sitio de transporte en una membrana o una enzima de reparación). A dosis bajas, el número de moléculas celulares reactivas residentes disponibles no cambia apreciablemente en función de la concentración del "ingreso". Por lo tanto, el número de colisiones relevantes y las tasas de las reacciones y reacciones laterales en la secuencia causal a bajas dosis deben ser funciones lineales directas de las cantidades de tóxico ingresado y sus derivados activados.
A dosis altas, sin embargo, puede haber una saturación de los procesos de detoxificación, activación o reparación a lo largo de la ruta de las transformaciones reactivas/carcinogénicas del ADN. A dosis altas que saturan estos procesos clave de detoxificación o reparación, existe el potencial para una dependencia cuadrática del riesgo con la dosis, en relación con la dosis entregada en una exposición simple. La saturación de múltiples procesos, incluyendo la posible estimulación de la replicación celular, compondrá a futuro la relación dosis-respuesta a estas dosis transicionales, resultando en una escalada ascendente muy abrupta del riesgo sobre un rango de exposición de dosis bastante estrecho.
El rol de los sistemas enzimáticos de detoxificación
El papel que pueden jugar en la toxicidad los sistemas enzimáticos saturables involucrados en la detoxificación es algo bien conocido. La alcohol deshidrogenasa (ADH) representa el ejemplo clásico de una enzima de detoxi-ficación que se satura fácilmente, resultando en una cinética de orden cero para el metabolismo del alcohol. Más allá de las implicancias legales y sociales de una ADH saturable, hay consecuencias toxicológicas e implicancias para la evaluación del riesgo en el caso de las enzimas de detoxificación saturadas. Ya en los años ochenta, Gillette discutió el papel de las transiciones del metabolismo dependientes de la dosis que llevaban a curvas dosis-respuesta no lineales (14). Planteó que las curvas dosis-respuesta no lineales existían y que asumir que todas las dosis-respuestas eran lineales basándose en los efectos de las dosis altas no era correcto. Los datos mecanísticos generados desde entonces apoyan su moción de que existen las curvas dosis-respuesta no lineales. Ciertamente, los sistemas enzimáticos que están involucrados en la detoxificación pueden saturarse, y como resultado, los efectos observados en animales expuestos a dosis muy altas pueden no reflejar lo que ocurre para dosis muy bajas. Es probable que la saturación de las enzimas de detoxificación resulte en toxicidad pero la transición no siempre es directa.
Los sistemas enzimáticos de la biotransformación de xenobióticos se clasifican como reacciones de fase I y de fase II. Las reacciones de fase I comprenden modificaciones que incrementan la solubilidad en agua, y las de fase II reflejan conjugación con sustancias mayores en preparación para la excreción en un medio acuoso. Estos tipos de reacciones pueden competir pero a menudo son secuenciales en la vida biológica de un xeno-biótico. Ejemplos de enzimas de reacción de fase I son las esterasas, las deshidrogenasas y las enzimas del citocromo P450 (CYP). Algunos ejemplos de reacciones de la fase II involucran conjugación con GSH, con ácido glucurónico, con ácido acético y con sulfato.
Aunque algunas isoenzimas del P450 están asociadas con la activación de xenobióticos a las formas activas finales de los tóxicos, también algunas están involucradas en la detoxificación. Sustancias como los alcanos halogenados, aminas aromáticas y heterocíclicas, la aflatoxi-na B1 y otras tantas son detoxificadas por las isoenzimas CYP2E1 y CYP1A2. La determinación de los parámetros cinéticos para estas enzimas indica que CYP1A2 y CY-P2E1 son saturables. Por ejemplo, los valores de Km y Vmáx para el metabolismo del diclorometano por el CYP2E1 indican que esta enzima tiene una alta afinidad por el haloalcano, pero una baja capacidad. De manera similar, hay evidencia que las reacciones de detoxificación de la fase II que involucran a las glutatión transferasas, las sulfotransferasas y la acetilación, son saturables. Aquellas sustancias que exhiben cambios en su toxicidad dependiendo de la dosis, debidos a la saturación de las enzimas de detoxificación caen dentro de una de estas tres categorías, dependiendo del camino metabólico y la actividad biológica del compuesto original:
1) Para tóxicos que son detoxificados por un camino simple a niveles bajos de dosis, la toxicidad aumenta desproporcionadamente a niveles altos de dosis a medida que la enzima de detoxificación se satura. La alcohol deshidrogenasa es un ejemplo clásico. Se han determinado los parámetros cinéticos para ratas y la ADH tiene una Vmáx de 52 mg/hora. La exposición de las ratas a concentraciones altas de etanol, donde la enzima está saturada, se asocia con numerosos efectos tóxicos (15).
El etanol es un tóxico de vida media relativamente corta, y que sufre un metabolismo intenso a especies reactivas (acetaldehído y radicales libres) que son realmente las responsables de los efectos dañinos. En este laboratorio, se utilizan frecuentemente dos modelos experimentales en rata, el de la exposición aguda vía oral (alimentación forzada en una dosis única) y un modelo de exposición repetida (de un mes de duración, mediante una alimentación oral que puede considerarse ad libitum, aunque no sea así estrictamente). El comportamiento de muchos parámetros es absolutamente distinto entre ambos modelos. En principio, puede decirse que en la intoxicación aguda los niveles plasmáticos y tisulares de acetaldehído se mantienen altísimos durante un lapso significativo, reflejando la saturación de los mecanismos de eliminación (fundamentalmente de la oxidación metabólica) en el animal. En cambio, en el modelo de exposición repetida prácticamente no se detecta la acumulación de acetal-dehído (los niveles se mantienen bajos debido a que el balance entre su formación y su oxidación juega a favor de la destrucción del metabolito). En cuanto a los efectos, no son los mismos o tienen la misma intensidad en un modelo o en el otro (por ejemplo los relacionados con la producción de estrés oxidativo o las alteraciones morfológicas observadas) (16-19). El caso del testículo de rata lo ilustra claramente (Fig. 3) (Fig. 4).

Figura 3. Niveles de etanol in vivo en ratas intoxicadas en forma aguda con etanol (19).

Figura 4. Niveles de acetaldehído in vivo en ratas intoxicadas en forma aguda con etanol (19).
Luego de la administración de la dosis única de etanol por vía oral, los niveles plasmáticos siguieron una curva con un máximo producido a una hora de la administración. En testículo se observó presencia del etanol con un máximo a una hora desde la administración, en cantidades comparables al hígado y descendiendo a niveles mínimos hacia las 24 horas. Por su parte, en el hígado se observó la mayor cantidad de etanol presente en los tiempos entre una y 3 horas, cayendo en forma gradual hasta las 24 horas luego de la administración. En el gráfico siguiente se expresa (sólo para los grupos de animales intoxicados) la concentración de etanol en unidades similares a las utilizadas en mediciones de rutina para humanos (g/L para alcoholemia). La mayor concentración medida en plasma se aproxima a 3000 pg/mL, lo que equivale a 3 g/L, una cantidad esperable para una intoxicación fuerte en humanos.
En estos experimentos se quiso estudiar si el acetal-dehído podía acumularse transitoriamente en el tejido testicular, respecto de los niveles plasmáticos luego de la administración de una dosis única de etanol. En plasma no se observaron niveles importantes de acetaldehído en ninguno de los tiempos medidos. En testículos se observó una acumulación de acetaldehído creciente con el tiempo, aunque el máximo de estos valores, a las 3 horas de la administración, fue menor que los valores medidos en hígado, donde se observaron niveles más altos. Posteriormente la concentración decreció, equilibrándose con los niveles plasmáticos (19).
2) Otro caso de una transición dependiente de la dosis a través de la saturación de un camino de detoxificación lo constituyen los sistemas de dos enzimas que compiten por el mismo sustrato; una activa la sustancia mientras que la otra la detoxifica (o hace a la sustancia inasequible para la activación). La dosis-respuesta para la toxicidad depende entonces de la relación Vmáx/Km de cada enzima involucrada en la disposición metabólica del compuesto químico. Este parece ser el caso de la aflatoxina B1, la cual es metabolizada por la isoenzima CYP3A4 a un exo-8,9-epóxido y por la CYP1A2 a un en-do-8,9-epóxido (20). El exo-epóxido está asociado con eventos genotóxicos y la toxicidad subsiguiente, pero el endo-epóxido parece no ser genotóxico. Así, la saturación de la CYP1A2 podría llevar el metabolismo hacia el epóxido más tóxico y teóricamente, a un riesgo de cáncer mayor (21).
3) La situación donde se verifica un cambio se-cuencial dependiente de la dosis, de detoxificación a activación, debido a la saturación de las enzimas de detoxificación. El diclorometano constituye un ejemplo de una transición de detoxificación a activación. El metabolismo del diclorometano a niveles bajos de exposición ocurre a través de la CYP2E1, lo cual resulta en la formación de monóxido de carbono, que se une a la hemoglobina para formar carboxihemoglobina. La enzima de oxidación CYP2E1 tiene una gran afinidad por el diclorometano pero tiene una baja capacidad y por lo tanto se satura fácilmente. Así, a medida que la exposición o la dosis se incrementan, la enzima CYP se satura fácilmente, cambiando el metabolismo al camino de la glutatión-S-transferasa (GST), que tiene una afinidad menor pero mayor capacidad para el diclorometano. El metabolismo del diclorometano vía GST resulta en un metabolito reactivo (22). Es a concentraciones de diclorometano por encima de las cuales ocurre este cambio que se observan los tumores en animales de laboratorio. En efecto, la CYP2E1 y la GST-T1 no compiten por el diclorometano debido a la diferencia en el Km, pero la saturación de la isoenzima CYP es una parte importante en la evaluación del riesgo (3)(23).
Factores que modifican el metabolismo de xenobióticos y su modulación por la dosis
En la evaluación del papel que juega la saturación enzimática en las transiciones dependientes de la dosis, hay factores modificadores adicionales que deberían considerarse. Uno es el rol de la inducción enzimática, otro son las diferencias entre las especies en la tasa metabólica (cinética) y otro son los polimorfismos genéticos. Es obvio que detrás de los efectos aparentes de la dosis sobre la respuesta estarán operando algunos de estos factores. Recíprocamente, puede concluirse que la influencia de estos factores no necesariamente será la misma, dependiendo de la dosis (Tabla II).
Tabla II. Ejemplos de diferencias entre especies en parámetros cinéticos.

Es algo conocido que algunas enzimas asociadas con el metabolismo de xenobióticos pueden inducirse para aumentar su actividad y/o cantidad. Así, algunas sustancias aumentan su propio metabolismo por exposición repetida. También es cierto que las enzimas pueden ser inducidas por sustancias no relacionadas, las cuales entonces pueden influir en la toxicidad del xenobiótico bajo estudio. Por ejemplo, las enzimas CYP2E1 son capaces de metabolizar algunos xenobióticos, incluyendo el diclorometano, varias N-nitrosaminas y el etanol. La exposición repetida de los animales (y de los humanos) al etanol induce la actividad de la 2E1 y el contenido de P450, lo que lleva a un aumento en el metabolismo del diclorometano y de varios carcinógenos. De este modo, la exposición repetida al etanol puede llevar a un incremento en el riesgo de la formación de carboxihemoglobina debido al metabolismo aumentado del diclorome-tano por la CYP2E1 o al aumento de la mutagenicidad de otros compuestos (24).
Se han demostrado diferencias entre las especies en la actividad específica, en los valores de Km y Vmáx, para las isoenzimas CYP y para las isoenzimas GST. Estas diferencias pueden tener un impacto significativo sobre la evaluación del riesgo. La tabla anterior muestra ejemplos de diferencias entre especies en parámetros cinéticos. Es claro entonces que la confianza total en los datos originados en animales de experimentación puede no ser apropiada para definir completamente el riesgo en humanos.
Otro factor modificador es el constituido por los polimorfismos genéticos de las enzimas. Se conocen muchos ejemplos de polimorfismos de las enzimas de detoxificación modulando tanto la toxicidad de xeno-bióticos como la actividad terapéutica de fármacos. En años recientes, los toxicólogos han apreciado mejor la diversidad genética de la población humana y cómo la genética influye en la evaluación del riesgo. Para algunos individuos, los polimorfismos llevan a un aumento en el riesgo debido a la pérdida de la capacidad de detoxificar, mientras que en otros disminuye el riesgo debido a la ausencia de la capacidad metabólica.
Agotamiento del cosustrato
La toxicidad de algunos xenobióticos es mediada por su reactividad química, ya sea la del compuesto original directamente o frecuentemente, la de los meta-bolitos de naturaleza electrofílica (25). Por lo tanto, la toxicidad global refleja un balance entre la activación (o ingreso, en el caso de xenobióticos inherentemente reactivos) y la detoxificación metabólica. Un mecanismo primario para la detoxificación de compuestos tóxicos es a través de la conjugación de las moléculas reactivas con los co-sustratos polares provistos por la célula (metabolismo de fase II). Biológicamente, esto sirve a dos propósitos. En principio, la conjugación enmascara al grupo funcional reactivo, reduciendo o eliminando su reactividad química y protegiendo el organismo contra la injuria tóxica. En segundo lugar, la conjugación generalmente incrementa la polaridad total y la solubilidad en agua de la molécula, facilitando el clearance desde el cuerpo a través de la excreción urinaria. La capacidad para detoxificar los compuestos reactivos por conjugación depende de varios factores, no siendo el menor de ellos la disponibilidad de los co-sustratos adecuados. Así, mientras que las concentraciones y/o síntesis de cosustrato pueden ser adecuadas para proteger contra las exposiciones de bajo nivel a tóxicos reactivos o pro-tóxicos, la administración del mismo compuesto en dosis altas puede llevar a una disminución rápida de la reserva de co-sustratos, produciendo una transición dosis-dependiente en la disposición metabólica del compuesto y en la no linealidad resultante en la curva dosis-respuesta. La hepatotoxicidad de una droga antipirética y analgésica, el acetaminofeno, sirve para ilustrar este concepto (2).
El acetaminofeno (paracetamol) no es tóxico en dosis terapéuticas, pero a dosis muy altas causa necrosis hepática severa, consecuencia de la producción exagerada de un metabolito reactivo. Este fármaco es meta-bolizado principalmente por la conjugación con ácido glucurónico y con sulfato. Un camino competitivo, pero de ordinario mucho menos relevante, involucra la oxidación mediada por el P450 a un metabolito altamente reactivo, la N-acetil-^benzoquinonimina (NAPQI, por su sigla en inglés). A dosis terapéuticas de acetaminofeno, los caminos de conjugación predominan y la pequeña cantidad de NAPQI producida es detoxificada efectivamente por conjugación con glutatión (GSH). A dosis altas, sin embargo, los cosustratos para los caminos de la sulfatación y la glucuronidización, 3'-fosfoadenosina-5'-fosfosulfato (PAPS) y UDP-ácido glucurónico (UDPGA) respectivamente, se consumen rápidamente causando un cambio hacia el metabolismo oxidativo. Esto a su vez resulta en la generación de cantidades anormalmente altas de NAPQI, llevando a la disminución de la reserva de GSH hepático.
Cuando la concentración del GSH hepático llega a un nivel crítico (aproximadamente 30-40% de lo normal) no habrá ya suficiente GSH para atrapar el NAPQI adicional. Como resultado de esto, hay un aumento abrupto de la unión covalente del NAPQI a blancos celulares relevantes y, consecuentemente, de la necrosis hepática en función de la dosis (Fig. 5). Este ejemplo demuestra cómo un patrón relativamente complejo de cómo se produce el agotamiento del cosustrato puede resultar en una no-linealidad marcada en la relación dosis-respuesta.
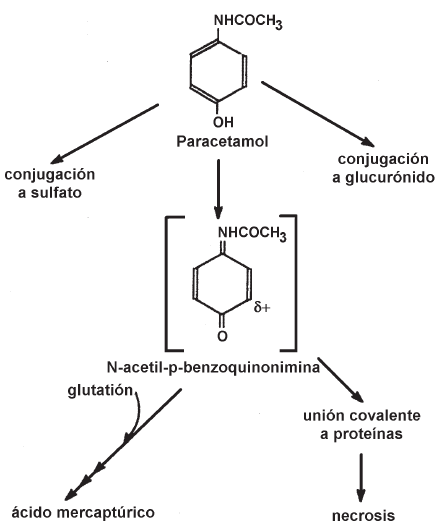
Figura 5. Vías metabólicas probables para la activación del paracetamol. El metabolito reactivo sería la N-acetil-p-benzoquinonimina. Esta especie puede reaccionar con glutatión para formar un conjugado o con grupos sulfhidrilo de proteínas. Alternativamente, el glutatión puede reducirlo a paracetamol, formándose simultáneamente glutatión oxidado.
El agotamiento de co-sustratos inducido por xenobióticos puede también ser de consecuencia significativa para otros procesos metabólicos que compiten por la misma cantidad de co-sustrato. En el feto en desarrollo, el sulfato es un importante co-sustrato en la biosíntesis de ciertos glicosaminoglicanos, los cuales son componentes de la matriz extracelular y son necesarios para el desarrollo adecuado del esqueleto y otras macromo-léculas relacionadas estructuralmente. La salicilamida es eliminada a través de la conjugación con el sulfato. A dosis altas, la salicilamida causa la disminución de los depósitos maternos y por ende fetales de sulfato, que podrían entonces no estar disponibles para la formación de glicosaminoglicanos. La pérdida de sulfato disponible en un feto creciendo rápidamente se manifiesta secundariamente con teratogénesis y crecimiento fetal disminuido.
Transformación/activación química
Algunos compuestos químicos ejercen sus efectos farmacológicos y/o toxicológicos mediante su transformación en otras especies. Una forma de biotrans-formación es la conversión a un metabolito activo, el cual puede causar una respuesta biológica. Uno de los modelos experimentales clásicos de toxicidad hepática, el tetracloruro de carbono, ilustra perfectamente esta situación (26). El etilenglicol es otro buen ejemplo de este fenómeno. Los estudios mecanísticos y farmaco-cinéticos muestran que un metabolito particular del etilenglicol, el ácido glicólico, exhibe una cinética no lineal, y demuestran una transición dosis-dependiente que puede llevar al desarrollo de toxicidad en animales bajo circunstancias específicas (2).
Los estudios, que determinan un amplio rango de puntos finales, han mostrado que el etilenglicol tiene una actividad tóxica intrínseca pequeña. El metaboli-to clave responsable por la mayoría de los efectos agudos del etilenglicol es el ácido glicólico. El etilenglicol es metabolizado enzimáticamente a glicolaldehído, el cual es luego oxidado rápidamente en ácido glicólico. La oxidación subsiguiente del ácido glicólico a ácido glioxílico es el paso limitante principal de la velocidad en la biotransformación (Fig. 6).

Figura 6. Metabolismo de etilenglicol. El NADH producido es utilizado en la producción de lactato. ADH: alcohol deshidrogenasa; ALDH: aldehido deshidrogenasa; LDH: lactato deshidrogenasa, GAO: ácido glicólico oxidasa; AO: aldehido oxidasa.
En estudios con animales usando dosis altas de etilenglicol, la saturación de la oxidación del ácido glicólico lleva a la acumulación de este metabolito tóxico. Parece que los factores para exceder esta transición son: (a) una dosis alta de etilenglicol, (b) una ruta con absorción rápida (por ejemplo, oral), y (c), la administración a una tasa rápida (por ejemplo, por sonda). Todos estos factores convergen para causar niveles muy altos de etilenglicol, el cual es convertido rápidamente en ácido glicólico. Los altos niveles resultantes de glicólico saturan su conversión enzimática a ácido glioxílico, llevando a un aumento desproporcionado en los niveles del ácido glicólico.
Si estas exposiciones a dosis altas de etilenglicol son relevantes para los humanos es algo cuestionable. Para exposiciones ocupacionales, de consumo u otras típicas humanas, la gran capacidad de los sistemas de metabolización disponibles para oxidar el etilenglicol completamente sin siquiera aproximarse a la saturación de la oxidación del ácido glicólico, se piensa que efectivamente previenen la posibilidad de efectos inducidos por el etilenglicol en humanos.
Homeostasis alterada
Todos los organismos vivientes, desde las bacterias hasta los mamíferos, mantienen un ambiente interno bien controlado para sostener su bioquímica básica y su biología molecular. Estos ambientes celulares consisten en concentraciones de varios componentes requeridos para convertir las señales entrantes en niveles apropiados de macromoléculas y concentraciones necesarias de varias moléculas pequeñas, asociados con las operaciones normales del funcionamiento celular requeridas para la supervivencia y la reproducción.
La homeostasis se refiere a la capacidad de los organismos vivientes para mantener el ambiente interno adecuado de cara a los cambios en las condiciones externas. La homeostasis no es una propiedad estática de los organismos. En lugar de esto, representa interacciones específicas de los componentes regulatorios que sirven para controlar los cambios en el ambiente interno a través de un mecanismo de retroalimentación negativa o para reconocer y responder a nuevas situaciones, por activación del circuito genético normalmente silencioso en el genoma.
En una célula funcionando normalmente, las concentraciones de algunos componentes están reguladas por procesos de control tales como la activación alostérica y la inhibición de caminos por el producto final. Estos procesos interactivos están diseñados para mantener concentraciones de los constituyentes celulares (aminoácidos, nucleótidos, etc.) dentro de límites estrechos saludables. La relación usual para el control por retroalimentación negativa del circuito electrónico tiene una región donde las alteraciones en el ingreso llevan desde muy pequeños incrementos en la respuesta hasta rangos donde posteriores aumentos en la señal sobrepasan los mecanismos de control y producen alteraciones marcadas en la salida. Dichas curvas son comunes en la descripción del ruido en un sistema de sonido a medida que el poder de salida aumenta. Las transiciones de la toxicidad dosis-dependientes con varios compuestos pueden ocurrir bajo situaciones donde las tasas de la dosis sobrepasan las respuestas homeostáticas o donde la inducción de una respuesta homeostática, tal como la inducción enzimática para el clearance de un xenobiótico, lleva a respuestas tisulares asociadas con proliferación celular e inducción tumoral. A continuación se describen varios ejemplos de transiciones dosis-dependientes asociadas con respuestas homeostáticas.
Nutrientes esenciales. Es sabido que varios nutrientes esenciales y vitaminas son tóxicos a dosis altas. Estos compuestos presentan regiones de deficiencia, regiones de adecuación y regiones de exceso en relación con sus relaciones dosis-respuesta. Por ejemplo, el manganeso se requiere para el desarrollo normal y mantenimiento de la salud en los adultos. Varias enzimas importantes requieren Mn como cofactor y la forma catiónica divalente está involucrada en el metabolismo de los ácidos nucleicos. En exceso, sin embargo, el Mn está asociado con manganismo, un síndrome neurológico que produce desórdenes del movimiento que tienen algunas similitudes con los desórdenes del movimiento del tipo del Parkinson (3). Otros metales esenciales también son tóxicos a dosis altas, incluyendo el cobre y el zinc. Los metales no son los únicos nutrientes esenciales que tienen transiciones dosis dependientes desde las dosis asociadas con esencialidad hasta aquellas asociadas con toxicidad. Algunas vitaminas como la A y la D, pueden acumularse en el cuerpo llevando a la toxicidad. La vitamina A es un requerimiento absoluto para el desarrollo correcto; sin embargo, en exceso causa una variedad de respuestas teratogénicas dependiendo de la dosis y del tiempo de gestación cuando se la administra (27). El oxígeno es esencial para la vida; sin embargo, las presiones aumentadas de O2 en cinco veces desde un 20% hasta atmósferas de oxígeno puro llevan a toxicidad pulmonar y del sistema nervioso. En cada caso, con manganeso, con vitamina A o con oxígeno, varios mecanismos mantienen las concentraciones apropiadas o manejan adecuadamente los metabolitos potencialmente adversos. Para el Mn, las concentraciones sistémicas están reguladas por la absorción dosis-dependiente y por la excreción biliar (2) (3).
Hormonas. La concentración de las hormonas en el cuerpo es mantenida por interacciones entre los órganos productores de hormonas (las gónadas, corteza adrenal, tiroides, etc.) y el eje hipotalámico-hipofisa-rio en el cerebro. Para la mayoría de estas hormonas naturales, las concentraciones están reguladas por retroalimentación negativa entre las concentraciones de hormona circulante y la liberación de factores estimulantes, como la hormona folículo estimulante (FSH), la hormona luteinizante (LH), la hormona estimulante de la tiroides (TSH), etc., desde la pituitaria. El ciclo reproductor femenino normal también depende de respuestas de retroalimentación positiva que regulan las oleadas de LH en el cerebro al estra-diol circulante. Las transiciones dosis-dependientes en toxicidad pueden ocurrir en el tejido testicular si la homeostasis es sobrepasada por un gran ingreso de testosterona u otros agonistas androgénicos. A medida que las concentraciones circulantes de testosterona se incrementan, cae la liberación de FSH y LH (a través de la reducción de la liberación de GnRH desde el hipotálamo). Las concentraciones altas de agonistas androgénicos en la dieta pueden disminuir la liberación de FSH desde la pituitaria, llevando a condiciones con concentraciones excesivas de andrógeno sistémico que están asociadas paradójicamente con atrofia testicular. Estos efectos ocurren a causa de la falla de los mecanismos homeostáticos para el control de las hormonas para manejar un ingreso muy elevado de hormonas o agonistas de ellas.
Eventualmente, estrategias similares a las mencionadas para la evaluación de riesgo de nutrientes traza pueden tener que aplicarse para compuestos que actúan como agonistas o antagonistas hormonales.
Mecanismos de reparación
Hay algunos ejemplos de cambios dependientes de la dosis en la reparación del ADN. Los ejemplos mejor descriptos se dan para los agentes alquilantes simples.
Algunos estudios demostraron claramente que una dosis única de dimetilnitrosamina, cubriendo cinco órdenes de magnitud, produce una cantidad de aducto N7-metilguanina (N7-MG) en forma lineal, pero un aumento agudo sub-lineal en la O6-metilguanina (O6-MG) (28). Esto ocurre en el punto de la curva dosis-respuesta donde la saturación de la proteína de reparación del ADN, la O6-metilguanina-ADN metiltransferasa (O6-MGMT) se satura. Estos dos aductos se forman (cuando se los produce químicamente) a un cociente constante de ~10 N7-MG : 1 O6-MG. Sin embargo, el aducto O6-MG es reparado rápidamente en el hígado, de tal forma que el cociente resulta aproximadamente 100:1 a dosis bajas, pero se torna en 10:1 a dosis que saturan la enzima O6-MGMT. Además, la misma proteína de reparación O6-MGMT que remueve el aducto O6-MG puede reparar la O4-metiltimina, un aducto que se forma en un 1/16avo de la cantidad de O6-MG y se repara con mucha menos eficiencia, llegando a concentraciones de estado estacionario casi iguales al aducto O6-MG después de dos a cuatro semanas de dosis diarias de la nitrosamina (Figura 7).

Figura 7. Relación dosis-respuesta entre N7-MG y O6-MG en hígados de ratas expuestas a una única dosis de dimetilnitrosamina. La línea entera representa la N7-MG, la línea punteada en la primera figura representa la cantidad teórica de O6-MG, y los círculos representan la cantidad real de O6-MG. La cantidad real de O6-MG es menos que la cantidad teórica a dosis menores debido a la reparación por la O6-MGMT. El segundo gráfico demuestra que la transición entre reparación efectiva de la O6-MG y la saturación de esta reparación resulta en un incremento de la pendiente en la dosis molecular de O6-MG. Referencia: (2).
Los datos obtenidos con la dietilnitrosamina son todavía más informativos para revelar las transiciones dosis-dependientes en la reparación del ADN. Mientras la O6-etilguanina (O6-EG) es formada químicamente a una tasa que es ocho veces mayor que la O4-etiltimidina (O4-ET), luego de cuatro semanas de exposición a 40 ppm de dietilnitrosamina, se formarán 50 partes más de O4-ET. Este es el resultado de la reparación eficiente de la O6-EG, al igual que de la inducción de la O6-MGMT que sigue a la exposición a la nitrosamina (29). Finalmente, cuando se examina la dosis molecular de aductos de ADN con la dietilnitrosamina, en un rango de 0,4 a 100 ppm en el agua corriente durante cuatro semanas, se identifica una transición dosis dependiente adicional.
La O2-etiltimina (O2-ET) muestra que satura su reparación a 40 y 100 ppm, resultando en un incremento adicional de este aducto, relativo al aducto O4-ET. Así, a 40 ppm de dietilnitrosamina, ocurre la inducción de la O6-MGMT, llevando a una remoción muy eficiente de la O6-MG (resultando en cantidades menos que proporcionales de este aducto, hasta el punto que no puede ser detectado), acumulación lineal de O4-ET y saturación de la remoción de O2-ET (llevando a cantidades más que proporcionales de este aducto) en el hígado del mismo animal (Figura 8).

Figura 8. Acumulación de 02-ETy 04-ET después de exposición continua de ratas por 28 días a 0,4, 1; 4; 10; 40 y 100 ppm de dietilni-trosamina en el agua de bebida. Las concentraciones de 02-ETy 04-ET están expresadas en función de la exposición a la dietilnitrosamina. Referencia: (2) (29).
Estos ejemplos ponen en evidencia varios fenómenos que merecen considerarse en la evaluación del riesgo tóxico. Primero, lo que se ve en los experimentos de dosis única puede no representar lo que sucede con dosis continuas o incluso intermitentes. Segundo, la reparación del ADN puede ser inducida y también puede saturarse. La transición dosis-respuesta para una proteína de reparación puede ser diferente de la de otra. Hay numerosos ejemplos de inducción de la reparación del ADN.
Esto se tornará aún más evidente con el advenimiento de las investigaciones toxicogenómicas. Por ejemplo, se demostró un aumento de la expresión de los genes para las enzimas oxidativas de reparación del ADN en ARN hepático de ratas y ratones tratados con prolife-radores de peroxisomas (30). En las figuras anteriores, puede verse claramente la saturación de dos caminos reparadores diferentes. Además, debe reconocerse que la mayoría de los mecanismos de reparación del ADN utilizan múltiples enzimas. En el caso de la reparación por escisión de bases, se requieren glicosilasas, AP en-donucleasas, polimerasa p y ligasa, para la reparación completa. Es concebible entonces que los desequilibrios en dicho camino resulten en la inducción o saturación de la reparación que podría provocar la remoción del aducto pero también en la acumulación de una lesión subsiguiente, tal como una rotura de cadena en el ADN.
Flujo sanguíneo y limitación de la difusión
Cuando se considera el movimiento de los xenobióti-cos desde la sangre arterial hacia el parénquima tisular, es conveniente definir por separado los compartimentos de sangre y parénquima tisular. La sangre arterial lleva sustancias dentro del compartimiento de sangre tisular a una velocidad (cantidad/tiempo) determinada por el flujo sanguíneo tisular (volumen/tiempo) y por la concentración del xenobiótico (cantidad/volumen). En cualquier punto en el tiempo, la sangre tisular tiene una cierta concentración (cantidad/volumen) del xenobiótico.
Para el caso donde el xenobiótico se mueve desde la sangre tisular hacia el parénquima por difusión, la velocidad de movimiento (cantidad/tiempo) puede estar limitada tanto por la velocidad del flujo sanguíneo dentro del compartimiento de la sangre tisular como por la tasa de difusión desde la sangre tisular al parénquima. Para moléculas grandes (por ejemplo, la dioxina TCDD), la velocidad de difusión desde la sangre al parénquima es relativamente lenta y la absorción está limitada por la difusión. Con las moléculas más pequeñas, la tasa de difusión es relativamente alta y la velocidad de absorción está limitada por el flujo (por ejemplo, el furano).
La capacidad tisular para el clearance metabólico (cantidad/tiempo) puede ser mayor que la velocidad a la cual el xenobiótico llega al compartimento sanguíneo tisular.
Con esta restricción, y cuando la absorción del xenobiótico está limitada por el flujo, la velocidad del metabolismo del xenobiótico también está limitada por el flujo. Se demostró que las velocidades de oxidación del furano en los hígados de ratones, ratas y humanos están limitadas por el flujo durante exposiciones de 4 horas hasta 300 ppm (31).
Una consecuencia importante del clearance metabó-lico bajo condiciones limitadas por el flujo es que la variación interindividual en la capacidad para el clearance metabólico (es decir, en el caso del furano, la variación en la CYP2E1) no lleva a una variación en la velocidad de clearance metabólico. Sólo cuando la capacidad para el clearance metabólico es menor que la tasa de ingreso del tóxico la tasa del clearance está limitada por la capacidad metabólica.
IMPLICANCIAS PARA LA EVALUACIÓN DEL RIESGO Y EL DISEÑO DE ESTUDIOS TOXICOLÓGICOS
Una pregunta que podría surgir luego de este análisis es: ¿para qué sirve todo este conocimiento de las variables que modulan la toxicidad de algo?
La respuesta es que las transiciones dependientes de la dosis en los mecanismos de toxicidad pueden impactar sobre la evaluación del riesgo para cualquier efecto adverso. Actualmente, las evaluaciones de riesgo para efectos no-cáncer generalmente establecen un nivel de efectos adversos no observados (en inglés, NOAEL) o un límite por debajo del mínimo detectable (BMDL) asociados con un nivel bajo de respuesta especificado y divide estos valores por factores de incertidumbre (seguridad) para llegar a una dosis diaria aceptable (ADI) o a una dosis de referencia (RfD), que se presume que resultará en un riesgo insignificante para la salud. En la ausencia de NOAEL, puede usarse el nivel más bajo de efectos adversos observados (LOAEL). Los factores de incerti-dumbre (seguridad) se emplean para dar cuenta de la extrapolación de los resultados en animales a humanos, sensibilidades variables a los compuestos químicos entre individuos y cuando es necesario, extrapolar a partir de las dosis que muestran efectos, al igual que otras consideraciones como la pertinencia de la base de datos. Este proceso supone la existencia de una dosis umbral (una forma de transición dosis dependiente) debajo de la cual la toxicidad observable no ocurre. Se supone generalmente que la ADI o la RfD están debajo de la dosis umbral y así se considera que presentan un riesgo insignificante para la salud. Un defecto de este proceso es que un riesgo importante (por encima del 20%) puede pasar desapercibido para ciertos tipos de experimentos, debido a la pérdida de poder estadístico (32). Derivar un ADI o un RfD de un NOAEL inapropiado puede no producir un riesgo insignificante. El reemplazo del NOAEL por el BMDL permite que la ADI o la RfD se calculen a partir de un nivel de respuesta conocido. En cualquier caso, el proceso real para ajustar un ADI o un RfD generalmente supone la existencia de una transición dosis respuesta, es decir, una dosis umbral, pero no intenta describir o utilizar información sobre los mecanismos de toxicidad dosis respuesta para reducir la incertidumbre en el establecimiento de la ADI o la RfD.
En el pasado, una presunción general por defecto en las evaluaciones del riesgo de cáncer era que cualquier exposición a un genotóxico puede incrementar el riesgo de cáncer incluso a dosis muy bajas. Esto ha llevado al uso de la extrapolación lineal conservadora a cero a partir de los resultados en animales, a menudo de dosis altas. La definición de dosis baja frecuentemente es problemática y este proceso no tiene en cuenta la inducción de la detoxificación o los procesos competidores que pueden reducir o eliminar los riesgos de cáncer a dosis bajas. Más recientemente, las guías de evaluación de riesgo de cáncer propuestas (33) proveen la oportunidad de considerar el margen de exposición entre el BMLD y los niveles de exposición humana anticipados en lugar de la extrapolación lineal de casos donde los datos ayuden a la previsión de una curva dosis respuesta no lineal en el rango de las dosis bajas. Así, la demostración de una transición dosis-dependiente para los procesos carcinogéni-cos podría tener una profunda influencia en la elección de los procedimientos de evaluación del riesgo empleados y los estimados subsecuentes de riesgo de cáncer o la determinación de una ADI o una RfD.
Consideraciones similares deben extraerse para los puntos finales de toxicidad no-cáncer.
Pregunta: ¿Qué información se les puede proveer a los evaluadores de riesgo para demostrarles que están ocurriendo las transiciones dependientes de la dosis? ¿Cuáles son los datos mínimos requeridos para demostrar que están ocurriendo las transiciones dependientes de la dosis?
La EPA fue comisionada para establecer prioridades y tomar decisiones, usando la "mejor información científica disponible" (34). La responsabilidad primordial de la agencia, sin embargo, es asegurar la protección de la salud pública. Así, la agencia toma lo que cree que son decisiones protectoras de la salud cuando la duda científica es significativa. Por ejemplo, la presunción por defecto presumiblemente protectora de la salud de la linealidad de las dosis bajas es usada a pesar de la incertidumbre significativa acerca de la forma de la curva dosis-respuesta para un agente carcinogénico (35). Por lo tanto, un desafío en la investigación toxicológica aplicada a la evaluación del riesgo es reducir la incertidumbre a un grado tal que los tomadores de decisiones de todos los sectores estén cómodos usando datos empíricos en lugar de presunciones por defecto.
Pregunta: ¿Cómopuede reducirse la incertidumbre?
Varias actividades interdependientes son relevantes para lograr esta meta. Los diseños experimentales deben ser adecuados para demostrar el significando tanto biológico como estadístico y para sostener el desarrollo de modelos computacionales. No es suficiente la simple demostración de la no linealidad de una curva dosis-respuesta.
El mecanismo que genera la no linealidad debe ser caracterizado y desarrollarse el modelo computacional del mecanismo. Se evalúan entonces la incertidumbre asociada con los datos y el modelo computacional asociado. Hay disponibles varios enfoques para el análisis de la incertidumbre, algunos con enfoques cuantitativos (36) y que ponderan el valor del análisis de la información (37) (38). La U.S. EPA otorga una fuerte confianza sobre el análisis cuantitativo de la incertidumbre en la forma de "informes narrativos" que describen las fortalezas y debilidades de la información disponible (35).
La necesidad de consenso es la base del trabajo experimental, el desarrollo de modelos y el análisis de incertidumbre. No puede esperarse que los datos meca-nísticos afecten la evaluación del riesgo a menos que se haya alcanzado un grado razonable de consenso acerca del mecanismo y las implicancias del mismo en la evaluación del riesgo.
Puede asumirse que el diseño experimental es adecuado para demostrar el significado tanto biológico como estadístico.
Los reguladores se convencen más probablemente de usar los datos generados en el laboratorio sobre mecanismos de acción tóxica cuando existe un consenso científico acerca de esos datos y sus implicancias para la incertidumbre. El consenso se define como una opinión colectiva o un acuerdo general. Es difícil definir el consenso cuantitativamente; por lo tanto, sólo pueden hacerse declaraciones generales acerca del consenso respecto a las no linealidades toxicológicas de dosis-respuesta. La aparición de un manuscrito revisado por pares en la literatura indica que varios revisores encontraron este trabajo digno de publicación. Sin embargo, los resultados que no están en completo acuerdo unos con otros pueden sobrevivir independientemente al proceso de revisión y aparecer en la literatura. Es así que se han publicado varios modelos PBPK para el butadieno en la literatura y se convocó una reunión de los autores de estos artículos para tratar de reconciliar las diferencias entre los modelos (39-41). Sin embargo, este esfuerzo no fue exitoso, y la EPA quedó con un mensaje mixto de la literatura. Entonces, una simple publicación revisada por pares e incluso varios artículos que tratan del mismo tópico (por ejemplo, los modelos PBPK para el butadieno) pueden fallar en reflejar un acuerdo general. La moraleja es que algo más allá de la publicación en la literatura revisada por pares puede ser necesario para alcanzar el consenso, por lo menos en situaciones parecidas a la descripta antes para el butadieno.
La Justicia trata a menudo con casos de daño tóxico donde se evalúan reclamos de efectos adversos para la salud relacionados con una exposición química. De alguna forma, estas evaluaciones son paralelas a la evaluación de los datos tóxicos de las agencias regulatorias. En ambas situaciones, el énfasis está en la extrapolación, donde "extrapolación" significa una inferencia estimada a partir de los datos disponibles. El daño tóxico involucra la consideración de si las bases de datos de la evaluación del riesgo y toxicológica son consistentes con un efecto (reclamado) para la salud. La evaluación del riesgo en una agencia regulatoria incluye típicamente (como mínimo) extrapolaciones entre especies y de dosis altas a bajas. En ambos escenarios, estas actividades son realizadas, con un grado variado de éxito, por grupos de individuos con distinto tipo de experiencia en las cuestiones técnicas y con familiaridad con los datos. Estos problemas han sido tomados en cuenta por el sistema legal. En Daubert vs. Merrell Farmacéutica (2), la Corte Suprema de los EE.UU. proveyó una guía para la evaluación de la información científica en la forma de cinco criterios:
(1) Si las teorías y técnicas empleadas por el experto científico han sido probadas.
(2) Si han estado sujetas a revisión por sus pares y publicadas.
(3) Si las técnicas empleadas por el experto tienen una tasa de error conocida.
(4) Si están sujetas a estándares que regulan su aplicación.
(5) Si las teorías y técnicas empleadas por el experto gozan de aceptación amplia. Daubert vs. Dow Merrell Farmacéutica suplantó a Frye vs. Estados Unidos (2), el cual requería más simplemente la "aceptación general" de la ciencia relevante. Los méritos relativos de Daubert y Frye son el sujeto del debate actual y están más allá del alcance de este análisis (42-44). Solamente el quinto criterio de Daubert se refiere directamente a la cuestión del consenso, y, sobre todo, Daubert es más útil para la consideración de si existen los ingredientes necesarios para el consenso. EPA también ha estudiado la cuestión de si existen los ingredientes necesarios para el consenso (35). EPA ha adaptado los criterios de Bradford-Hill para causalidad (45) para usarlos en la evaluación del riesgo de cáncer:
(1) Relación temporal
(2) Consistencia
(3) Magnitud de la asociación
(4) Gradiente biológico
(5) Especificidad de la asociación
(6) Coherencia
Daubert vs. Dow Merrell Farmacéutica (2) y la U.S. EPA (35) juntas proveen una abundancia de criterios por los cuales puede evaluarse el potencial de la información científica para sostener un consenso, o incluso la existencia del consenso en si mismo.
Hay actividades de construcción del consenso disponibles que van mucho más allá de la publicación en la literatura revisada por pares. El Instituto de Ciencias de la Salud y Ambientales (HESI, ILSI) convocó un panel experto para considerar, entre otras cosas, si la base de datos del cloroformo era consistente con una dosis-respuesta no lineal para sus efectos carcinogénicos (46). Las conclusiones de este panel tienen posiblemente más credibilidad que las publicaciones individuales revisadas por pares que el mismo panel consideró.
Otro enfoque consiste en involucrar a los contribuyentes en el desarrollo de la evaluación del riesgo basado en el mecanismo. Una evaluación de riesgo de cáncer para el formaldehído (Centros de Investigación para la Salud del Instituto de Toxicología de la Industria Química, CIIT) usó este enfoque (47). Mientras que el grueso del trabajo técnico de esta evaluación fue realizado por el CIIT, el trabajo fue guiado por un comité director con representantes de la EPA, la industria química y el CIIT. Los encuentros regulares de este comité durante el desarrollo de la evaluación ayudaron a asegurar que las cuestiones de importancia eran identificadas y estudiadas, y generalmente ayudaron a obtener el apoyo de los contribuyentes. La culminación del proceso fue una reunión revisada por pares con un objetivo concreto convocada por la EPA de los EE.UU. y Salud de Canadá. Salud de Canadá publicó desde entonces un documento consistente con la evaluación (Formaldehído. Reporte de la evaluación de la lista de sustancias prioritarias. ISBN 0-662-29447-5. Cat. N° En40-215/61E) y la comisión alemana MAK también promulgó una nueva regulación del formaldehído basándose en parte en la evaluación (Deutsche Forschungsgemeinschaft. Lista de valores MAK y BAT 2000. Comisión para la Investigación de los Peligros para la Salud de los Compuestos Químicos en el Área de Trabajo. Reporte N° 36, Wiley-VCH). Hoy el formaldehído es considerado un carcinógeno humano por la Agencia Internacional para la Investigación del Cáncer (48).
En resumen, los datos mecanísticos de cualquier tipo, incluyendo los datos que describen las transiciones dosis-dependiente, son usados más probablemente para la evaluación del riesgo y la regulación cuando la reducción de la incertidumbre puede demostrarse y comunicarse efectivamente. La efectividad aumenta hasta el grado de que se logran criterios como aquellos listados en Daubert vs. Dow Merrell Farmacéutica (2) y en la U.S. EPA (35). Paneles de expertos bien constituidos (por ejemplo, los de HE-SIILSI) han considerado estos aspectos, y todos los contribuyentes apropiados han sido involucrados en el proceso de evaluación del riesgo desde sus etapas más tempranas (46).
Una implicancia de este análisis es que una única publicación revisada por pares puede caer bien cerca de la marca cuando lo que se intenta es usar la ciencia para afectar la regulación. El desafío puede ser visto en términos probabilísticos. La probabilidad de que los datos mecanísticos influyan en la evaluación del riesgo aumenta hasta el punto en que haya múltiples publicaciones revisadas por pares, preferiblemente no todas de los mismos autores, que los paneles de expertos hayan considerado la literatura y que los contribuyentes hayan estado involucrados en el desarrollo de la evaluación del riesgo.
PERSPECTIVAS SOBRE LAS CUESTIONES DE SELECCIÓN DE DOSIS EN EL DISEÑO DE ESTUDIOS TOXICOLÓGICOS
Los distintos tipos de transiciones en el mecanismo de la toxicidad podrían observarse a través de un amplio rango de concentraciones de exposición, o dosis administradas. Una cuestión crítica es si los efectos observados son relevantes a las exposiciones en el mundo real de los humanos o si son artefactos del régimen de dosificación. Estas consideraciones son por consiguiente críticas para la selección de las dosis usadas en el bioensayo.
Se han descripto cinco principios para la selección de dosis en los bioensayos crónicos (49). Los principios generalmente se discuten en el contexto del diseño de bioensayos de cáncer, pero los principios son realmente para el diseño de cualquier estudio toxicológico. Estos principios incluyen: (a) aplicación de principios toxicológicos firmes, sosteniendo el uso de dosis en aumento, lo cual maximiza la sensibilidad para permitir la detección de los efectos, pero no dentro de rangos que violen dichos principios y que lleven a resultados que son inapropiados para la evaluación del riego en humanos; (b) aplicación de un diseño apropiado de estudio junto con datos mecanísticos y otra información toxicológica cuando se seleccionan las dosis del bioensayo, reconociendo que los peligros identificados pueden ser dosis-dependientes; (c) consideración de los niveles de exposición humana potenciales, modo/ mecanismo de acción, toxicocinética y otros datos, particularmente cuando se seleccionan las dosis medias y las más bajas, una de las cuales puede (o no) tener un nivel de efecto adverso no observado (NOAEL); (d) que los puntos finales generalmente evaluados en los estudios pre-crónicos y que ayudan a definir la forma de la curva dosis-respuesta, junto con otra información relevante, deberían ser considerados cuando se diseñan los estudios a largo plazo; y (e) que deben considerarse los factores fisicoquímicos para minimizar o evitar los efectos adversos nutricionales, organoléptico-físicos e irritantes. Un principio abarcador establecido por el International Life Sciences Institute-Risk Science Institute (ILSI-RSI) es que la ciencia "que pesa", más que la tradición o la adherencia rutinaria a las normas (a pesar de los requerimientos regulatorios) es lo que debe aplicarse para la selección de la dosis.
Queda claro de estos principios y otras discusiones sobre el diseño de bioensayos que los procesos dinámicos y cinéticos que son dependientes de la dosis deben ser tenidos en cuenta tanto dentro del diseño como de la interpretación de los bioensayos (50) (51).
La selección de las dosis de los bioensayos es, sin embargo, un ejercicio prospectivo. Esto es, la selección de las dosis está basada en información anterior, y esta información puede ser óptima o no. Por esta razón, se recomienda fuertemente en todos los estadios del proceso de evaluación la consideración de las transiciones dosis-dependientes en toxicocinéticas y toxicodinámicas, comenzando con estudios agudos y finalizando en el bioensayo crónico.
La toxicocinética, quizás el menos apreciado de los puntos finales de rutina en el ensayo de toxicidad de pesticidas y en la industria química, puede evaluarse como un punto final adjunto en todos los estadios de la evaluación de la seguridad. Las mediciones analíticas simples de la concentración plasmática luego de dosis únicas o repetidas pueden ayudar a determinar si está ocurriendo la saturación de los procesos involucrados en la absorción, distribución, metabolismo y eliminación del compuesto original. Los parámetros cinéticos tales como la vida media plasmática y el área normalizada de la dosis por debajo de la curva se obtienen con una mínima colección de muestras y pueden servir como indicadores de las cinéticas no lineales bajo las condiciones del bioensayo.
La evidencia de la no linealidad podría sugerir saturación de los caminos fisiológicos y bioquímicos que es probable que resulten en una interpretación confusa de los hallazgos del bioensayo y compliquen la extrapolación del riesgo a dosis bajas.
La identificación de los metabolitos principales en el plasma y las excretas es también de valor para la determinación de las transiciones dosis-dependientes en el mecanismo. Los caminos metabólicos de baja capacidad pueden saturarse a dosis altas causando un cambio en el perfil de los metabolitos. Dicha observación sugiere no sólo un cambio en la cinética sino que podría señalar también un giro en la dinámica, que es la introducción de un nuevo camino de activación metabólica. Un ejemplo bien estudiado es el del cloruro de metile-no, donde a dosis bajas predomina un camino de eliminación metabólica dependiente del citocromo P450. A altas concentraciones de exposición, este camino se satura y la eliminación metabólica es derivada hacia un camino dependiente del glutatión. El camino glutatión-dependiente es tóxico y está asociado con cáncer de hígado. Es importante notar que los avances significativos en la tecnología analítica, más especialmente en la espectrometría de masas en tándem/HPLC, han reducido significativamente el costo, la dificultad técnica y los requerimientos de muestra necesarios para adquirir estos datos. Esto es, ahora pueden obtenerse datos toxicocinéticos de gran valor para las consideraciones mecanísticas y cinéticas en la selección de la dosis en los bioensayos y su interpretación.
De igual modo, puede obtenerse importante información que señale cambios dosis-dependientes en la toxicodinámica en los estudios pre-crónicos que son útiles para la selección de dosis en el bioensayo. Sin embargo, la detección de cambios en la toxicodinámi-ca requiere biomarcadores de respuesta más sensibles que los tradicionales puntos finales de toxicidad tales como peso del órgano, la histopatología, y la química clínica. Las mediciones de la proliferación celular en el órgano blanco (por ejemplo, bromodeoxiuridina o antígeno nuclear de proliferación celular) han ganado una amplia aceptación como marcadores de un cambio hacia mecanismos de carcinogenicidad liderados por la proliferación. La inducción enzimática hepática, cuan-tificable por inmunofluorescencia, el ensayo enzimático funcional o las técnicas histoquímicas, constituyen ejemplos de un cambio dinámico que indica cambios dosis-dependientes en el mecanismo. Recientemente, ha habido un interés creciente en la determinación de cambios hormonales durante los estudios precrónicos (52-55). Dichas evaluaciones poseen la capacidad de mostrar cambios en las respuestas dependientes de andrógenos o estrógenos en los tejidos endocrinos o de órganos reproductores. El desarrollo de biomarcadores de respuesta más efectivos que sean útiles para determinar la transición dosis-dependiente en el mecanismo de acción, al igual que determinar la distinción entre una respuesta normal o adaptativa reversible o compensatoria y una respuesta adversa, es un área que necesita cada vez mayor atención.
En conclusión, la determinación de las transiciones dosis-dependientes en el mecanismo de acción es una consideración importante en todos los bioensayos, desde el diseño de los estudios agudos hasta los crónicos. Deberían incluirse dentro de estos diseños elementos de evaluaciones toxicocinéticas y toxicodinámicas para mejorar la información disponible cuando se diseñen los bioensayos subsiguientes extendidos o crónicos. Estos datos, junto con los datos similares recolectados durante el bioensayos crónico, resultarán en una interpretación más fundamentada de los hallazgos del bioensayo en relación con la relevancia humana y con la extrapolación del riesgo a dosis bajas.
IMPLICANCIAS PARA LA SELECCIÓN DE ESPECIES EN EL DISEÑO DE ESTUDIOS DE BIOENSAYO
Como se discutió arriba, las transiciones dependientes de la dosis en el mecanismo de acción dependen de factores cinéticos y dinámicos. Siendo estos factores ampliamente diferentes entre las especies de testing y los humanos, entra en discusión la relevancia de la respuesta que es dependiente de estos factores. Este aspecto tan importante vinculado con la selección de los datos que son relevantes a la evaluación del riesgo tóxico excede el espacio de esta discusión pero debe suponerse que los factores cinéticos y dinámicos relevantes son operativos en especies de testing y en humanos (y en las sub-poblaciones humanas) y que las diferencias entre ellos son cuantitativas, más que cualitativas.
Debido a que los factores cinéticos y dinámicos son inherentemente dependientes de los procesos fisiológicos y bioquímicos que, finalmente, están gobernados al nivel del genoma, es razonable esperar diferencias cuantitativas entre especies en las dosis por encima de las cuales ocurren dichas transiciones. El conocimiento previo de estas diferencias podría influir en la elección del modelo animal usado en un bioensayo de toxicidad o en la elección de los niveles de dosis que finalmente se compararán para las exposiciones humanas conocidas o esperadas.
Es necesaria una guía para dirigir la evaluación del riesgo de sustancias químicas que presentan transiciones dependientes de la dosis en su mecanismo de acción para apoyar el compromiso de usar la mejor ciencia posible en el proceso de evaluación del riesgo. Puede ser posible obtener cálculos directos del riesgo a partir de datos experimentales para una función dosis-respuesta bien definida en la región de las dosis bajas. Sin embargo, todavía sería necesaria esa guía para permitir la extrapolación a humanos y a las diferencias entre individuos. Presumiblemente, la comprensión mejorada de los mecanismos de toxicidad y el acompañamiento de las funciones de dosis-respuesta debería sostener el uso de factores de incertidumbre menores y, de ese modo, la derivación de ADIs o RfDs más altos.
Supóngase que la curva dosis-respuesta provee una medida de la incidencia o la severidad de un efecto adverso para la salud como función de la dosis. Una pendiente mayor de la curva dosis-respuesta indica un efecto adverso mayor para la salud por unidad de dosis. Para una transición dosis-dependiente de la toxicidad, existe un rango de dosis de transición donde la pendiente de dosis-respuesta aumenta o disminuye notablemente, produciendo por eso una dosis-respuesta semejante a un umbral o en forma de palo de hockey. Se discute debajo el caso especial de la dosis-respuesta en forma de U, que puede indicar un rango de riesgos para la salud reducido. Con una combinación de información biológica adecuada y datos experimentales, debería establecerse una dosis de transición específica para servir como reemplazo para el NOAEL, el LOAEL o el BMDL (límite por debajo del mínimo detectable) en las evaluaciones del riesgo. Para una dosis respuesta semejante a umbral, el avance es que la dosis de transición se aproximaría generalmente a la dosis más alta asociada con un riesgo insignificante, a lo sumo. Para una dosis-respuesta en forma de "palo de hockey", el establecimiento de una dosis de transición permitiría la extrapolación a dosis bajas a partir de exposiciones a dosis altas empleando factores de incertidumbre mejor informados.
Para el caso especial de la dosis-respuesta en forma de U, hay un rango de dosis en el cual la incidencia o severidad de los riesgos para la salud está disminuida. En este caso, hay una dosis equivalente al cero (ZED) donde la respuesta es igual a la de los controles. En esta situación, las dosis bajas no sólo se permitirían sino que realmente serían beneficiosas. Una ADI o una RfD podrían ser tan grandes como el ZED sin aumentar los riesgos para la salud por encima de los niveles de base. Incluso si se empleara una extrapolación lineal conservadora, la extrapolación al ZED resultaría en una reducción más rápida en el riesgo estimado que la extrapolación a cero. Establecer la porción en forma de U de una dosis-respuesta en la región de la dosis baja puede resultar posiblemente en valores de ADIs o RfDs con niveles de riesgo que son menores que los de fondo. Claramente, no hay ganancia en establecer ADIs o RfDs por debajo de la dosis que se considera que representa un riesgo insignificante.
Los bioensayos con animales son realizados típicamente con dosis altas para provocar la toxicidad potencial con números de animales relativamente bajos. El propósito principal de dichos estudios es discriminar la toxicidad, y se le presta poca atención a examinar los efectos en el rango de las dosis bajas. Esto puede ser adecuado para regular los riesgos para las funciones dosis-respuesta no semejantes a umbral. Sin embargo, para las relaciones dosis-respuesta que muestran un umbral, y particularmente para las curvas dosis-respuesta en forma de U, es necesaria una descripción de la función dosis-respuesta para predecir más exactamente los riesgos asociados con exposiciones a dosis bajas. Esto necesitará la inclusión de unas pocas dosis bajas en los bioensayos. Esto también requerirá generalmente el uso de mediciones de toxicidad más sensibles, distintas a la incidencia de mortalidad, cáncer o defectos de nacimiento.
Conclusiones
Una transición dependiente de la dosis es una variación o cambio con el aumento de la dosis en factores clave cinéticos y/o dinámicos subyacentes que influyen en el mecanismo responsable de la toxicidad observada, resultando en un cambio en la relación de la tasa de respuesta en función de la dosis (Figura 1). Debido a que el factor definido puede ser cinético o dinámico, el uso de modelos cinéticos basados fisiológica y biológicamente es importante para identificar los factores clave que influyen en una transición particular. Existen numerosos ejemplos de transiciones dosis-dependientes para efectos tanto cáncer como no cáncer en animales experimentales. Algunos de estos han sido investigados rigurosamente y atribuidos a las transiciones entre distintos mecanismos de toxicidad. En humanos, se supone que, en ausencia de una evidencia en contra, el modo de acción es cualitativamente el mismo que el demostrado experimentalmente, y que el conocimiento adquirido desde los animales de laboratorio es directamente transferible para anticipar las transiciones dosis-dependientes cuando se calculan los riesgos para la salud en humanos.
La identificación de un rango de dosis asociadas con la transición es clave para definir apropiadamente el rango de dosis más bajo para la extrapolación entre especies, género y estadios de vida. Además, la identificación de una transición dosis-dependiente permite más confianza en la extrapolación más allá del rango de dosis ensayado y en el cálculo de los márgenes de exposición. Desafortunadamente, las estrategias actuales de ensayo probablemente no revelarán las transiciones en la relación dosis-respuesta, dejando sin contestar la pregunta crítica de donde, dentro de la relación dosis-respuesta, ocurre la transición con respecto a otros puntos críticos de referencia (por ejemplo, exposición humana real o el NOAEL derivado experimentalmente).
Para identificar mejor las transiciones y el rango de dosis sobre el cual ellas ocurren, los estudios de toxicología deberían enfatizar la colección de datos mecanísticos firmes (por ejemplo, cambios bioquímicos, datos de proliferación celular, etc.). A medida que las bases mecanísticas de las transiciones dosis-dependientes se van poniendo en claro, aumentará la confianza en el uso de la experimentación in vitro y otras alternativas de ensayo, resultando en una reducción del número de animales necesarios para las evaluaciones toxicológicas tradicionales. Además, la caracterización del mecanismo de toxicidad en animales revelará biomarcadores de respuesta útiles, los cuales son esenciales para anticipar puntos de partida en la relación dosis-respuesta humana. Esos biomarcadores deberían estar vinculados con un efecto adverso observado y con uno que sea considerado confiable en humanos (lo que se denomina un "biomarcador puente").
En un esquema de ensayo toxicológico mejorado, son importantes los ensayos de sensibilidad suficiente para detectar la evidencia de una transición con confianza estadística. También debe admitirse que una hipótesis del modo de acción servirá mejor como base de una estrategia de ensayo dirigida. Entre las posibles modificaciones que pueden hacerse al diseño del estudio toxicológico tradicional están las siguientes: perfil de toxicidad in silico, recolección de datos fisicoquímicos, recolección de información toxicocinética a múltiples niveles de dosis y en el testing con dosis repetidas y el uso de animales transgénicos y otros modelos de enfermedad. Además, la selección de dosis debería enfatizar la región de transición de la curva dosis-respuesta para definir mejor el rango de exposiciones para el cual la extrapolación no lineal puede ser más crítica.
La aplicación del concepto de transiciones dosis dependientes en la evaluación del riesgo requiere una comprensión mejorada de la exposición. La caracterización de las transiciones no será tan importante si solo se la presenta en el contexto del peligro. Por lo tanto, es esencial caracterizar la relación entre la dosis aplicada, la interna y la dosis en el sitio blanco para comprender completamente su significado.
En conclusión, la mayoría de los científicos y tomadores de decisiones de todos los sectores concuerdan en que las evaluaciones de riesgo deberían estar basadas en la "mejor ciencia disponible". Debido justamente a que existen algunos ejemplos de desviaciones de una relación lineal cuando uno explora el rango total de la curva dosis respuesta, la consideración de las transiciones dependientes de la dosis en el mecanismo de toxicidad es un ejemplo obligado de cómo integrar la "mejor ciencia" dentro del proceso de toma de decisiones.
La Toxicología ha ingresado en una era excitante donde el uso de información más sofisticada y basada biológicamente está comenzando a informar el proceso de evaluación del riesgo y la regulación de los agentes tóxicos. Claramente, la identificación y descripción cuantitativa de las transiciones dosis-dependientes en los mecanismos de toxicidad es una parte importante de la caracterización de los riesgos para la salud.
CORRESPONDENCIA
Dr. Gerardo D. Castro Centro de Investigaciones Toxicológicas CEITOX-UNIDEF, MINDEF-CONICET Juan B. de La Salle 4397 B1603ALO VILLA MARTELLI, provincia de Buenos Aires E-mail: gcastro@citedef.gob.ar
Referencias bibliográficas
1. Castro GD. Epidemiología molecular: La contribución de la química a la prevención de enfermedades de origen ambiental. En: Galagovsky L, directora. Química y Civilización. Buenos Aires: Asociación Química Argentina; 2011. p. 121-30. [ Links ]
2. Slikker W Jr, Andersen ME, Bogdanffy MS, Bus JS, Cohen SD, Conolly RB, et al. Dose-dependent transitions in mechanisms of toxicity. Toxicol Appl Pharmacol 2004; 201 (3): 203-25.
3. Slikker W Jr, Andersen ME, Bogdanffy MS, Bus JS, Cohen SD, Conolly RB, et al. Dose-dependent transitions in mechanisms of toxicity: case studies. Toxicol Appl Pharmacol 2004; 201 (3): 226-94. [ Links ]
4. Hattis D. A pharmacokinetic/mechanism-based analysis of the carcinogenic risk of ethylene oxide. MIT Center for Technology, Policy and Industrial Development, Cambridge, Report No. CTPID 87-1, August 1987.
5. De Silva PE. Determination of lead in plasma and studies on its relationship to lead in erythrocytes. Br J Ind Med 1981; 38 (3): 209-17. [ Links ]
6. Chamberlain AC. Prediction of response of blood lead to airborne and dietary lead from volunteer experiments with lead isotopes. Proc R Soc Lond B Biol Sci 1985; 224 (1235): 149-82.
7. Gehring P, Watanabe PG, Park CN. Risk of angiosarcoma in workers exposed to vinyl chloride as predicted from studies in rats. Toxicol Appl Pharmacol 1979; 49 (1): 15-21. [ Links ]
8. Maltoni C, Lefemine G. Carcinogenicity bioassays of vinyl chloride: current results. Ann N Y Acad Sci 1975; 246 (1): 195-218.
9. Belinsky SA, Walker VE, Maronpot RR, Swenberg JA, Andersen ME. Molecular dosimetry of DNA adduct formation and cell toxicity in rat nasal mucosa following exposure to the tobacco specific nitrosamine 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone and their relationship to induction of neoplasia. Cancer Res 1987; 47 (22): 6058-65.
10. Kaden DA, Call KM, Leong PM, Komives EA, Thilly WG. Killing and mutation of human lymphoblast cells by af-latoxin B1: evidence for an inducible repair response. Cancer Res 1987; 47 (8): 1993-2001. [ Links ]
11. Swenberg JA, Gross EA, Martin J, Popp JA. Mechanisms of formaldehyde toxicity. En: Gibson JE, editor. Formaldehyde Toxicity. Washington (DC): Hemisphere Publishing Corporation; 1983. p. 132-47.
12. Hurtt ME, Thomas DA,Working PK, Monticello TM, Morgan KT. Degeneration and regeneration of the olfactory epithelium following inhalation exposure to methyl bromide: Pathology, cell kinetics, and olfactory function. Toxicol Appl Pharmacol 1988; 94 (2): 311-28.
13. Bithell JF, Stiller CA. A new calculation of the carcinogenic risk of obstetric Xraying. Stat Med 1988; 7 (8): 857-64. [ Links ]
14. Gillette JR. Problems in risk assessment. J Environ Sci Health A 1982; 17 (4): 553-8.
15. Bevan C. Monohydric alcohols C1 to C6. En: Bingham E, Cohrssen B, Powell CH, editors. Patty's Toxicology. Volume 6. 5th ed. New York: Wiley; 2001. p. 382-94.
16. Castro GD, de Castro CR, Maciel ME, Fanelli SL, de Ferreyra EC, Gómez MI, et al. Ethanol-induced oxidative stress and acetaldehyde formation in rat mammary tissue: potential factors involved in alcohol drinking promotion of breast cancer. Toxicology 2006; 219 (1-3): 208-19.
17. Castro GD, Delgado de Layño AM, Fanelli SL, Maciel ME, Díaz Gómez MI, Castro JA. Acetaldehyde accumulation in rat mammary tissue after an acute treatment with alcohol. J Appl Toxicol 2008; 28 (3): 315-21. [ Links ]
18. Díaz Gómez MI, de Castro CR, Fanelli SL, Quintans LN, Costantini MH, Castro JA, et al. Biochemical and ultrastructural alterations in the rat ventral prostate due to repetitive alcohol drinking. J Appl Toxicol 2007; 27 (4): 391-8.
19. Quintans LN. Bioactivación del etanol en el testículo de rata y su rol en la toxicidad reproductiva en el alcoholismo (tesis doctoral). San Martín: Universidad Nacional de San Martín; 2008.
20. Guengerich FP, Johnson WW, Shimada T, Ueng YF, Yamazaki H, Langou Jt S. Activation and detoxification of aflatoxin B1. Mutat Res 1998; 402 (1-2): 121-8.
21. Ueng YF, Shimada T, Yamazaki H, Guengerich FP. Oxidation of aflatoxin B1 by bacterial recombinant human cytochrome P450 enzymes. Chem Res Toxicol 1995; 8 (2): 218-25. [ Links ]
22. Gargas ML, Clewell III HJ, Andersen ME. Metabolism of inhaled dihalomethanes in vivo: Differentiation of kinetic constants for two independent pathways. Toxicol Appl Pharmacol 1986; 82 (2): 211-23.
23. Jonsson F, Johanson G. A Bayesian analysis of the influence of GSTT1 polymorphism on the cancer risk estimate for dichloromethane. Toxicol Appl Pharmacol 2001; 174 (2): 99-112. [ Links ]
24. Díaz Gómez MI, Valles E, Fanelli SL, de Layño AM, Castro GD, Castro JA. Alcohol induction of liver nuclear ethanol and N-nitrosodimethylamine metabolism to reactive metabolites. Teratog Carcinog Mutagen 2002; 22 (2): 139-45.
25. Castro JA, Castro GD. Treatment of chemically induced free radical mediated cell injury. Anal Soc Cient Argent 1997; 227: 37-50.
26. Castro JA, Castro GD. Carbon tetrachloride. En: Sipes IG, Mac Queen CA, Gandolfi AJ, editors. Comprehensive Toxicology. Volume 9. Amsterdam: Elsevier Science; 1997. p. 251-71.
27. Meltzer HM, Aro A, Andersen NL, Koch B, Alexander J. Risk analysis applied to food fortification. Public Health Nutr 2003; 6 (3): 281-91. [ Links ]
28. Pegg AE, Hui G. Formation and subsequent removal of O6-methylguanine from deoxyribonucleic acid in rat liver and kidney after small doses of dimethylnitrosamine. Biochem J 1978; 173 (3): 739-48.
29. Swenberg JA, Dyroff MC, Bedell MA, Popp JA, Huh N, Kirstein U, et al. O4-Ethyldeoxythymidine, but not O6-ethyldeoxyguanosine, accumulates in DNA of hepa-tocytes of rats exposed continuously to diethylnitrosa-mine. Proc Natl Acad Sci USA 1984; 81 (6): 1692-5.
30. Rusyn I, Denissenko MF, Wong VA, Butterworth BE, Cunningham ML, Upton PB, et al. Expression of base excision repair enzymes in rat and mouse liver is induced by peroxisome proliferators and is dependent upon carcinogenic potency. Carcinogenesis 2000; 21 (12): 2141-5.
31. Kedderis GL, Held SD. Prediction of furan pharmacoki-netics from hepatocyte studies: comparison of bioacti-vation and hepatic dosimetry in rats, mice and humans. Toxicol Appl Pharmacol 1996; 140 (1): 124-30. [ Links ]
32. Leisenring W, Ryan L. Statistical properties of the NOAEL. Regul Toxicol Pharmacol 1992; 15 (2 Pt 1): 161-71.
33. U.S. Environmental Protection Agency (U.S. EPA). Guidelines for Carcinogen Risk Assessment. EPA/630/ P-03/001F. Washington (DC): U.S. Environmental Protection Agency; 2005.
34. U.S. Environmental Protection Agency (U.S. EPA). 2003-2008 EPA Strategic Plan: Direction for the Future. Washington (DC): U.S. Environmental Protection Agency; 2003.
35. U.S. Environmental Protection Agency (U.S. EPA). Proposed Guidelines for Carcinogen Risk Assessment. EPA/600/P-92/003C. Washington (DC): Office of Research and Development, U.S. EPA; 1996.
36. Bois FY. Statistical analysis of Clewell et al. PBPK model of trichloroethylene kinetics. Environ Health Perspect 2000; 108 (Suppl.): 307-16.
37. Brand KP, Small MJ. Updating uncertainty in an integrated risk assessment: conceptual framework and methods. Risk Anal 1995; 15 (6): 719-31. [ Links ]
38. Sielken Jr. RL. How to use both human and animal data in quantitative risk assessment. En: Graham JD, editor. The Role of Epidemiology in Regulatory Risk Assessment: Proceedings of the Conference on the Proper Role of Epidemiology in Risk Analysis, Boston, MA, USA, 1314 October 1994. New York: Elsevier; 1995. p. 105-23.
39. Johanson G, Filser JG. A physiologically based pharma-cokinetic model for butadiene and its metabolite butadiene monoxide in rat and mouse and its significance for risk extrapolation. Arch Toxicol 1993; 67 (3): 151-63.
40. Kohn MC. The importance of anatomical realism for validation of physiological models of the disposition of inhaled toxicants. Toxicol Appl Pharmacol 1997; 147 (2): 448-58. [ Links ]
41. Sweeny LS, Schlosser PM, Medinsky MA, Bond JA. Physiologically based pharmacokinetic modeling of 1,3-bu-tadiene, 1,2-epoxy-3-butene, and 1,2,3,4-diepoxybu-tane toxicokinetics in mice and rats. Carcinogenesis 1997; 18 (4): 611-25.
42. Deftos L. Many courts still Frye scientific evidence. Science 2002; 297: 1275.
43. Faigman DL. Is science different for lawyers? Science 2002; 297 (5580): 339-40.
44. Faigman DL. Response to Deftos. Science 2002; 297 (5580): 1275-6. [ Links ]
45. Rothman KT. Modern Epidemiology. Boston: Little, Brown and Company; 1986.
46. International Life Sciences Institute (ILSI). An Evaluation of EPA's Proposed Guidelines for Carcinogen Risk Assessment Using Chloroform and Dichloroacetate as Case Studies: Report of an Expert Panel. Washington (DC): ILSI HESI; 1997.
47. CIIT Centers for Health Research. Formaldehyde: hazard characterization and doseresponse assessment for carcinogenicity by the route of inhalation, (revised edition). Research Triangle Park, NC: Chemical Industry Institute of Toxicology; 1999.
48. I nternational Agency for Research on Cancer (IARC). Formaldehyde, 2-Butoxyethanol and 1-tert-Butoxypro-pan-2-ol. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Volume 88. Lyon: IARC Press; 2006.
49. Foran JA. Principles for the selection of doses in chronic rodent bioassays. ILSI Risk Science Working Group on Dose Selection. Environ Health Perspect 1997; 105 (1): 18-20. [ Links ]
50. International Conference on Harmonisation (ICH). Guideline for industry. Dose selection for carcinogenici-ty studies of pharmaceuticals. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Federal Register. Volume 60. p. 11278-81. 1995.
51. International Conference on Harmonisation (ICH). Toxicokinetics: the assessment of systemic exposure in toxicity studies. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Federal Register. Volume 60. p. 11264-8. 1995.
52. Ashby J. Increasing the sensitivity of the rodent utero-trophic assay to estrogens, with particular reference to bisphenol A. Environ Health Perspect 2001; 109 (11): 1091-4.
53. De Vito M, Biegel L, Brouwer A, Brown S, Brucker-Davis F, Cheek AO, et al. Screening methods for thyroid hormone disruptors. Environ Health Perspect 1999; 107 (5): 407-15. [ Links ]
54. O'Connor JC, Frame SR, Ladics GS. Evaluation of a 15-day screening assay using intact male rats for identifying steroid biosynthesis inhibitors and thyroid modulators. Toxicol Sci 2002; 69 (1): 79-91.
55. O'Connor JC, Frame SR, Ladics GS. Evaluation of a 15-day screening assay using intact male rats for identifying antiandrogens. Toxicol Sci 2002; 69 (1): 92108. [ Links ]
Aceptado para su publicación el 30 de noviembre de 2012

