











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCTION
Since several decades, the alloy of U with Mo in the γ phase (bcc) has been proposed as a highdensity nuclear fuel [1], which increases burnup and favors non-proliferation. In this alloy, the fission of the uranium isotopes produces Xe and Kr at the rate of one gas atom for every four fission events [2]. Inert gas atoms have a strong tendency to precipitate in small bubbles due to their low solubility.
Up to moderate burnup, the nanobubbles (mainly of Xe) have a diameter of 30-40 Å and are not randomly distributed but aligned in an fcc superlattice of about 120 Å lattice constant, which is oriented parallel to the bcc lattice of UMo [3]. This Xe nanobubble superlattice resembles similar structures that form in other metals (Mo, W, Al, Cu, and many others [4]) under ion/neutron irradiation at particular conditions (temperature, dose).
Shenyang Hu et al. [2] pointed out three possible mechanisms as responsible for nanobubble superlattice formation: elastic interaction, one-dimensional migration of self-interstitial atoms, and dislocation punch out. Several theoretical works can be found in the literature [2,5-8] exploring such mechanisms mostly under the assumption that they may be due to overpressurized bubbles. Nevertheless, Salvato et al. [9] recently found that, at low burnup, i) nanobubble lattice forms preferentially at grain boundaries and extends to the grain interior with increasing irradiation dose, and ii) bubbles are extremely underpressurized, suggesting its void-like nature.
In order to shed some more light on the above questions, nanovoids morphologies and interactions are presently studied for the U-10wt%Mo alloy. The molecular dynamics technique, at T = 500 K (estimated working temperature in a nuclear reactor) and periodic boundary conditions in all directions, is chosen. Embedded atom method (EAM) interatomic potentials derived from the work of Smirnova et al. [10] for U-Mo system are employed. All simulations are performed with the LAMMPS code [11] while most of the visualization and analysis of results are made with OVITO [12].
METHODS AND RESULTS
Average atom potential
To avoid the limitations entailed by making the simulations at a given, fixed, component distribution of the alloy, the averaging method of Varvenne et al. [13] is used. The method allows obtaining an average atom (AA) potential starting from the original EAM interaction. The AA potential constitutes a reasonably faithful representation of the heterogeneous system, e.g. stabilizing the γ phase (bcc) only at relatively high temperatures (T > 400 K). Table 1 reports some of the structural and elastic properties presently calculated for the studied alloy using each potential. In what follows simulations performed with the AA potential are referred to as homogeneous (material), while those using the original one on purely random mixtures, as heterogeneous.

Void faceting
The simplest shape one can imagine for a void is a sphere. Following this naïve approach, a spherical void of r = 20 Å (1984 vacancies) is carved in a cubic heterogeneous cell of around 144 Å edge 22at% Mo (149216 atoms, equivalent to 10wt% Mo). The cell is run for 3000 ps at constant NPT at a high temperature (T = 800 K) in order to accelerate atomic movements, in particular, at the surface of the void. The resultant surface diffusion is not homogeneous and depends on position on the surface.
Displacements are limited almost exclusively to atoms located near <111> directions on the void surface, while atoms on or close to {110} planes hardly move (see Fig. 1a). At the end of the simulation, flat atomic surfaces on the {110} and {100} densest planes start to develop. Fig. 1b and 1c show two possible ideal Wulff (equilibrium) shapes as suggested by simulation results.

FIG. 1: Void morphology in U-10%wtMo. a) (100) projection of the bcc lattice with a void of r = 20 Å showing atomic displacements (black arrows) after 3000 ps at T = 800 K. Colored circles are U/Mo atoms. Traces of two {110} planes are also shown. Ideal Wulff shapes of the void: b) {100} truncated and c) regular rhombic dodecahedrons. Miller indices of the planes are indicated.
Assuming that voids adopt the perfect polyhedral shape of Fig. 1c, simulations of faceted voids of r = 15 Å (671 vacancies) are performed in a cell of 143 Å edge (147505 atoms) for both the heterogeneous and homogenous material to characterize the defect. This time, simulations are performed at constant NVT with T = 500 K and lattice parameters taken from Table 1. In the case of the heterogeneous system, 100 short runs of 25 ps with different random configurations are averaged out to obtain a representative energy. The first 10 ps in each run are discarded to avoid any contribution from transient states. The formation energy E f of a vacancy cluster is calculated as:

where E N+def is the energy of the cell with N atoms and the defect, and E a the corresponding energy per atom appearing in Table 1. The obtained values are E f =(182.7±0.3) eV for the heterogeneous and E f =(207.9±0.2) eV for the homogeneous system. The difference between potentials might be attributed to possible limitations of the AA model at finite temperatures [14], already suggested by Table 1. The same methodology was used by other authors [15] to model a 5-component alloy. In this case, atomic sizes of the components do not differ in more than 3% and the mixing enthalpies of all binary mixtures are less than 0.7 kJ/mol [16]. These characteristics make the alloy an almost ideal mix and, therefore, the AA approximation performs particularly well. On the contrary, in the present U-Mo alloy the atomic sizes of both species differ by more than 12% and the enthalpy of mixing is about 4 kJ/mol at 22at%Mo [17]. As a result, the U-Mo mixture has important structural distortions, which are absent in a homogeneous medium such as the AA. It is also worth noting that the AA is strictly a method for calculating the average energy at 0 K. At finite temperatures the (average) thermodynamic energy of the heterogeneous medium will not necessarily match the AA result [14]. However, we believe that presently the temperature effect is less important than that of size. We must keep in mind these limitations when using the AA method for the UMo alloy.
The observed facet development is consistent with the atomic plane densities and energies in this alloy. Surface energies calculated for the most compact planes for the homogeneous and the heterogeneous system are reported in Table 2. The first principles (FP) unrelaxed values of Mei et al. [18] are also reported for comparison. The lowest energy occurs for the {110} compact bcc planes. On the other hand, differences between surface energies for homogeneous and heterogeneous materials may help to understand the disagreement between the obtained values of E f .
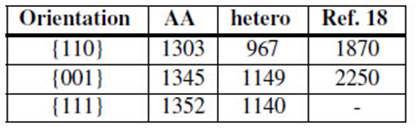
TABLA 2: Surface energies (mJ/m2) for the most compact planes in U-10wt%Mo at T = 500 K for the homogeneous (AA) and the heterogeneous (hetero) material. Also reported are the unrelaxed FP values from Ref. 18.
As a precedent in the literature, it is worth to mention the work of Miao et al. [19] in pure γU, showing that smallvacancy clusters remaining from a collision cascade adopt polyhedral shapes with {110} facets. Other authors indirectly suggest that faceting is related to the gas pressure exerted on the bubble surface [2,8,20,21]. Simulations performed by Shenyang Hu et al [2] in U-10wt%Mo with the same interatomic potential show that bubbles become faceted, but in {111} planes, when they emit dislocation loops by Xe overpressurization. On the other hand, Beeler et al. [8] found that faceting can result in an energy reduction in this alloy even at lower Xe pressures, but no comments are made on facet orientation. In transmission electron microscopy, faceting may be difficult to observe because out-of-focus conditions are required to measure bubble diameters and distances. On He+ ion implanted Fe-Cr alloys, Snoeck et al. [20] were able to reveal faceting in helium bubbles as small as 22 Å by means of electron holography. In the case of the UMo alloy, Leenaers et al. [21] found that the larger gas bubbles were faceted at high burn-up, when the superlattice ordering is lost.
In the following, it is considered that the equilibrium shape of a void in the UMo alloy is the polyhedron depicted in Fig. 1c and interactions for the U-10wt%Mo are reasonably well represented by the AA potential.
Two close voids
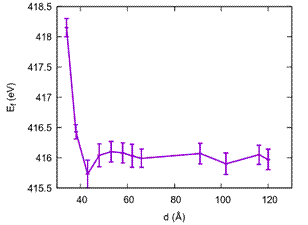
In order to study void interactions, two faceted voids with an average radius of r = 15 Å (671 vacancies) were created at distance d between their centroids in a large cell of 240 Å × 125 Å × 120 Å (185858 atoms) of homogeneous material. The bcc lattice was oriented such that one of the {110} facets of each void are directly facing each other along a <110> crystallographic direction. Distances were varied from almost contact up to 120 Å. As it is assumed that the bcc lattice do not change its bulk equilibrium volume by addition of these defects, simulations were performed at constant NVT. Fig. 2 shows the E f dependence on the distance d between voids, calculated by means of eq. 1. It can be seen a rather strong repulsion at very short distances and a rapid decay to a constant value corresponding to twice the formation energy for single void. In particular, the curve hardly shows any minimum that might set the superlattice parameter of experimental findings.
Void superlattices
To test for a preferred ordering, the fcc, bcc and simple cubic (sc) void superlattices are studied for the homogeneous material varying the first neighbor distance d 1nn . Spherical (r = 15 Å, 725 vacancies) and faceted voids (r = 15 Å, 671 vacancies) are NVT simulated at T = 500 K. In each superlattice, the simulation cell is chosen as the primitive supercell containing one void, except for bcc faceted case in which a cubic shape with two voids has been used (see Fig. 3). Spherical voids displayed some limited atomic movements while rearrangements of surface atoms on faceted voids were absent throughout the whole simulation.

FIG. 3: Simulation cells for each of the studied superlattices: a) fcc, b) bcc, c) sc. The a) and c) cells are primitive, containing one faceted void per cell, while the one for the bcc is twice the primitive cell and has two voids. Only atoms (gray circles) at the surface of the voids are shown.
Fig. 4 shows the dependence of the formation energy E f on d 1nn . Spherical voids do not seem to manifest any strong dependence. On the other hand, faceted voids in fcc arrangement show a behavior similar to that found for two close voids while no noticeable features can be seen for bcc and sc. Also, no preferred ordering is apparent.

SUMMARY AND CONCLUSIONS
Voids morphology and interactions are investigated for the U-10wt%Mo alloy by means of molecular dynamics at 500 K. A suitable average atom potential is used in the simulations, derived from the original potential of Smirnova et al. [10]. Spherical voids show inhomogeneous atomic movement at their surface, suggesting an evolution towards a different shape. On the other hand, {110} faceted voids are seen to be completely stable throughout all simulations, which can be explained by its lowest surface energy among all other orientations. Therefore, a {110} faceted void is proposed as the equilibrium Wulff shape. Interactions between voids of different morphologies are studied by varying distance and distribution in the bcc lattice. It is found that interactions are only present in faceted voids when two {110} facets oppose each other along a <110> direction at a distance no greater than a few atomic planes. This effect can be attributed to a short range surface-surface interaction. The current simulations suggest that elastic interactions are involved in neither ordering type nor superlattice size.

