










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.67 n.6-1 supl.1 Ciudad Autónoma de Buenos Aires 2007
Trastornos generalizados del desarrollo. Aspectos clínicos y genéticos
Víctor Ruggieri1, Claudia Arberas2
1Servicio de Neurología, Hospital de Pediatría Juan P. Garrahan;
2Sección Genética, Hospital de Niños Ricardo Gutiérrez, Buenos Aires, Argentina
Dirección postal: Dr. Victor Ruggieri. Servicio de Neurologia, Hospital de Pediatria Juan P. Garrahan, Combate De Los Pozos 1881, Buenos Aires, Argentina e-mail: vruggieri@intramed.net.ar
Resumen
Los Trastornos Generalizados del Desarrollo se expresan con compromiso en la socialización, trastorno en el desarrollo del lenguaje (verbal y no verbal) e intereses restringidos con conductas repetitivas. La frecuencia estimada en la población general es de 27.5/10.000. En nuestro trabajo analizamos los aspectos clínicos y genéticos de los TGD: Autismo, Síndrome de Asperger, TGD no Especificado, Síndrome de Rett y Trastorno desintegrativo de la niñez. Desde el punto de vista clínico jerarquizamos los aspectos conductuales para su reconocimiento. En los aspectos genéticos puntualizamos diversas entidades con las que se asocian consistentemente estos trastornos, denominados cuadros sindrómicos, (aproximadamente el 20% de los casos) y las bases genéticas actualmente propuestas para el 80% restante o formas no sindrómicas. El reconocimiento temprano de estos trastornos del desarrollo y el diagnóstico de una entidad específica asociada permiten un temprano y adecuado abordaje terapéutico, un correcto asesoramiento genético y un control evolutivo específico previendo posibles complicaciones relacionadas a la entidad de base. Finalmente, si bien las bases genéticas del autismo no están identificadas se han propuesto diversos genes candidatos ubicados en los cromosomas: 15q, 2q, 17q, 7q, 12q, y los relacionados al X, entre otros, los que son analizados en este trabajo y permitirán en un futuro cercano comprender mejor estos trastornos.
Palabras clave: Autismo; Síndrome de Asperger; Síndrome de Rett; Trastorno desintegrativo de la niñez
Abstract
Pervasive developmental disorders. Clinical and genetics aspects. Pervasive developmental disorders (PDD) encompass a heterogeneous group of children with deficits of verbal and non-verbal language, social communication, and with a restricted repertoire of activities or repetitive behaviours. The frequency in general population is considered 27.5/10,000. In this study, we analyzed the clinical and genetic aspects of Autism, Asperger Syndrome, PDD Not Otherwise Specified, Rett Syndrome and Childhood Disintegrative Disorder. We analyzed clinical, behavioural and neuropsychological features. We revised different medical genetics associated conditions and divided the genetics aspects of pervasive developmental disorders into two groups: Syndromic forms (around 20%) and non syndromic forms (currently proposed to be 80%). The early recognition of pervasive developmental disorders and the diagnosis of specific associated syndromes allow early therapy, correct genetic counselling, and follow up anticipating possible complications related to the entity. Finally, although the genetic bases of autism have not yet been identified, the following candidate genes have been proposed: 15q, 2q, 17q, 7q, 12q, and X related genes, among others; which are analyzed in this study and will allow a better understanding of these disorders in the future.
Key words: Autism; Asperger syndrome; Rett syndrome; Childhood desintegrative disorden.
Los Trastornos Generalizados del Desarrollo (TGD) se expresan por compromiso en la socialización, déficit del desarrollo del lenguaje (verbal y no verbal) e intereses restringidos con conductas estereotipadas.
En este trabajo analizaremos los aspectos clínicos y genéticos del Autismo, el Síndrome de Asperger, Trastorno Generalizado del Desarrollo no especificado, el Trastorno Desintegrativo de la niñez y el Síndrome de Rett, todos ellos incluidos dentro de la clasificación del Manual de Diagnóstico y Estadístico de los Trastornos Mentales número IV (DSM IV)1.
En primer lugar abordaremos el Autismo, el Síndrome de Asperger y el Trastorno Generalizado del Desarrollo No Especificado como un continuo de los trastornos profundos del desarrollo, con variaciones en sus aspectos cognitivos y conductuales que permite su identificación en el DSM IV1.
Luego analizaremos el Síndrome de Rett, como una entidad específica, con sus variaciones, y finalmente el trastorno desintegrativo de la infancia, un cuadro poco frecuente que debe ser tenido en cuenta especialmente considerando sus diagnósticos diferenciales.
Autismo
Desde las descripciones de Kanner en 19432 y Asperger en 19443 las interpretaciones sobre la génesis del Autismo pasaron desde la inconsistente y dañina culpabilización a los padres, hasta las actualmente aceptadas bases neurobiológicas y teorías neuro-psicológicas.
Podemos definirlo como: "un síndrome conductual de base biológica asociado a diversas etiologías"4.
Aspectos epidemiológicos
Se estima que el total de los Trastornos Generalizados del Desarrollo afectan aproximadamente a 27.5/10.000 personas5, mientras el autismo se estima en 10/10.0005 con una clara predominancia en varones siendo su relación 4/1.
Si bien el número de mujeres que lo padecen es estadísticamente menor, las afectadas tienen en general formas más severas.
En un interesante trabajo, Miles y Hilvanen en 20006, observaron que en autistas con dismorfias, examen neurológico anormal con o sin malformaciones del sistema nervioso central en la Resonancia Magnética de Cerebro (RM) la relación varón/mujer era de 1.7/1.
Cuando el examen neurológico era normal, y no había dismorfias la relación V/M era de 7/1.
Mientras que en las formas más puras, sin alteraciones en el examen neurológico ni dismorfias, y sin alteraciones estructurales demostrables en la Resonancia Magnética dicha relación era de 23/1. Indicando la existencia de determinantes ambientales en el primer grupo, comprometiendo casi de igual modo a varones que a mujeres y factores genéticos de importancia en el último grupo.
Aspectos conductuales
La ausencia de un marcador específico hace que el reconocimiento del cuadro autista dependa exclusivamente de la identificación de sus aspectos conductuales.
Se trata de un síndrome caracterizado por un severo trastorno en la socialización, compromiso en la comunicación, trastornos en el desarrollo del lenguaje, conductas repetitivas e intereses restringidos.
Socialmente son niños aislados, desinteresados por el entorno (muchas veces los padres refieren que "parecen sordos"), con pobre integración con sus pares, no tienen un juego simbólico, no juegan adecuadamente con los juguetes sino que muchas veces los investigan o los utilizan para hacer filas o torres, no conocen reglas de juego, su contacto visual es disperso y su mirada es en general huidiza, no reconocen las expresiones faciales y tienen escasa atención compartida.
Las conductas disruptivas e inadecuadas, muchas veces presentes en estos niños, empeoran su compromiso social, y podrían ser generadas, entre otras causas, a partir de su pobre comprensión de situaciones, la débil memoria retrógrada, el no comprender futuros premios y la escasa flexibilidad.
Todos los autistas tienen compromiso del lenguaje tanto verbal como no verbal.
- Compromiso del lenguaje no verbal: se evidencia esencialmente, por escaso contacto visual, déficit de la atención compartida, déficit para discriminar expresiones faciales y carencia de gestos expresivos.
- Compromiso en el lenguaje verbal: Es aceptado que aproximadamente el 50% de los autistas no lo desarrollan y los restantes pueden tener diversas alteraciones, entre otras, síndrome semántico pragmático, hipo espontaneidad, respuestas con monosílabos, ecolalias, jerga, estereotipias verbales, hablar en tercera persona y déficit receptivo.
- Conductas repetitivas o estereotipadas, pueden ser motoras (Ej. aleteos, saltar, deambular sin un fin determinado, balanceo con el cuerpo, abrir y cerrar puertas, cortar papeles, etc.), sensoriales (Ej. tocar superficies, chupar objetos, etc.), sensitivas (Ej. mirar la luz, mirar correr el agua, escuchar música, observar girar un objeto, etc.).
No obstante, debe tenerse en cuenta que el espectro conductual y cognitivo es muy variable en cada niño, por lo cual el reconocimiento de fortalezas y debilidades será fundamental para el abordaje del mismo.
Si bien las personas entrenadas en su reconocimiento identifican rápidamente estas características, es importante tener en cuenta que diversas escalas de evaluación como el CARS7, ADI-R8, ADOS9 y clasificaciones como la del DSM-IV1 permiten un diagnóstico cognitivo y conductual adecuado.
¿Cuándo y por qué consultan los padres de un niño con autismo?:
Es aceptado en general, que los padres de niños autistas consultan por primera vez aproximadamente a los 18 meses, porque el niño presenta trastornos en el desarrollo de su lenguaje. En nuestra experiencia, si bien coincidió la edad promedio de la primer consulta a un médico, el motivo de la misma fue en un 24% trastorno del lenguaje y conducta, 22% trastorno en el desarrollo de lenguaje, 21% trastorno de conducta, 17% retraso madurativo global, 12% convulsiones y 4% por otras causas10.
Como vemos, un alto número de estos padres no habían consultado por el lenguaje solamente, haciéndose el espectro de consulta más amplio que lo habitualmente reportado.
Por otra parte, si analizamos el motivo de consulta de nuestro grupo, cuya edad promedio fue 5 años, solo el 12% concurrió con diagnóstico conductual de autismo. Este aspecto fue para nosotros preocupante, sabiendo la importancia del abordaje temprano e intensivo en esta entidad10.
Aspectos genéticos
Numerosos estudios han permitido en el curso de los últimos años reconocer los aspectos genéticos determinantes del autismo.
Estudios en gemelos muestran la alta heredabilidad de esta condición, que exhibe una concordancia entre el 36% y 95% entre aquellos monocigóticos o idénticos (que comparten el 100% del material genético), versus los gemelos no idénticos o dicigóticos (que comparten el 50% del material genético) que coinciden en un 3-6%11, 12, 13.
Una vez descartada alguna etiología específica, se estima que los riesgos de repetición en hermanos es del 2-8% (es decir 100 veces superior al riesgo de la población general), 4% de ellos muestra una alteración social severa, y un 5.3% otro TGD14.
Estos riesgos varían en función de otros aspectos como el sexo del probando, la coexistencia con dismorfias leves, y la presencia o no de otros familiares igualmente comprometidos.
Determinar la etiología subyacente resulta de significativa importancia por numerosas razones.
1. Permite establecer un diagnóstico, contribuyendo a predecir un pronóstico evolutivo con cierto grado de certeza.
2. Posibilita la estimación de los riesgos de recurrencia para futuros hermanos y demás miembros de la familia.
3. Permite la selección de estudios complementarios necesarios en la confirmación de un diagnóstico presuntivo, como en el adecuado manejo evolutivo de patologías específicas.
4. Contribuye en la orientación del abordaje terapéutico.
5. Posibilita la contención emocional de los padres, frente a una patología crónica, invalidante, y de gran impacto afectivo-emocional.
Usualmente un 10% de los pacientes puede tener un diagnóstico etiológico presuntivo solamente mediante la evaluación dismorfológica y el relevamiento de los antecedentes familiares y personales.
En el 10-30% de los casos el diagnóstico etiológico es asequible con la utilización de otras técnicas complementarias10, 15-16.
Estudios a realizar: Una detallada historia clínica con un pormenorizado examen físico llevado a cabo por personal médico entrenado en dismorfología, estudios de citogenética preferentemente con técnicas de alta resolución de bandas, y en ocasiones con técnicas de marcación especial mediante sondas específicas (FISH), estudios moleculares específicamente orientados hacia una patología en particular, y la evaluación del SNC mediante estudios por imágenes (que permiten detectar alteraciones estructurales aun en ausencia de signos neurológicos), constituyen los estudios que se requieren para completar la evaluación de un paciente con esta patología15.
Actualmente, el correcto abordaje de estas entidades ha permitido diferenciar dos grandes grupos16, 17:
GRUPO 1) Formando parte de la expresión de Síndromes o entidades conocidas (estos son el 16-20% de los casos): S. Cromosómicos, S. Monogénicos y S. teratogénicos.
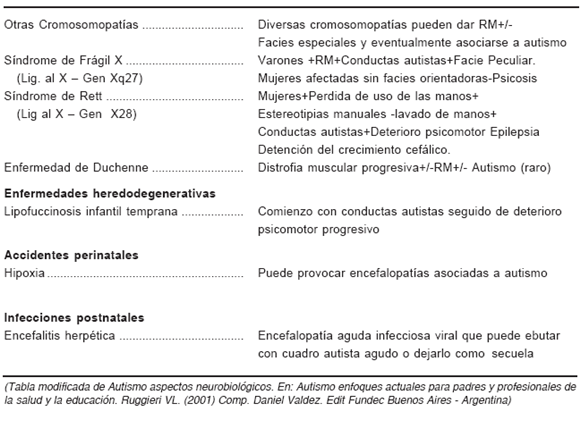
En la Tabla 1 realizamos una reseña orientadora de entidades neurológicas asociadas con autismo, algunas de las cuales desarrollaremos más adelante.
TABLA 1. - Entidades neurológicas asociadas a autismo

GRUPO 2) Formas no sindrómicas que representan casi el 80% restante cuya etiología obedece al efecto combinado de múltiples genes o etiología poligénica. Se analizarán ambos grupos en forma independiente:
Grupo 1: Formas sindrómicas
Síndromes reconocidos:
1. A) Cromosómicos: tanto aberraciones numéricas como estructurales han sido reconocidas como causantes de cuadros malformativos con retardo mental y autismo.
Aberraciones numéricas:
Síndrome de Down o trisomía del par 21 presenta una coincidencia con autismo del orden del 5-10%, lo que representa un riesgo 25 veces superior al de la población general18.
Síndrome 47, XYY reconocido ya en 1968, posee una prevalencia de 1: 1000 RN varones19, 20. Si bien esta cifra es importante existe un sub-diagnóstico de esta entidad por la falta de elementos de diagnóstico fácilmente detectables en la primera infancia. Los jóvenes y adultos presentan una talla final que supera en 10-12 cm. los valores normales. La presencia de dificultades en el aprendizaje es detectada en la escuela primaria, especialmente en la lecto-escritura. Severos trastornos emocionales y sociales incluyendo el Síndrome de Asperger ha sido reportado 21.
Síndrome de Turner o 45, X0 que muestra en un tercio de los casos claro perfil compatible con el Espectro autista22, 23.
Todas estas situaciones requieren un abordaje terapéutico especial, diferente al propio de la patología en sí mismo, por lo que su completa identificación es fundamental a los efectos de lograr un adecuado manejo del paciente.
Síndrome de Pallister Killian o Tetrasomía 12p, estos niños con un fenotipo claramente identificable por el ojo entrenado en dismorfología, presentan retardo mental, usualmente profundo, un patrón de comportamiento que encuadra perfectamente en el espectro autista; entidad esta que solamente puede reconocerse mediante estudios citogenéticos en cultivo de tejidos distintos a los linfocitos sanguíneos (de donde usualmente desaparecen antes del nacimiento)24.
Aberraciones estructurales:
Síndrome de Angelman (SA) (MCK #105830): producido en un 70% de los casos por deleción de la región crítica en el cromosoma 15q13 de origen materno, 7% por Disomía uniparental (DUP) paterna, 3% por defectos en la zona específica de impronta genómica ubicada en esa región y 11% otras mutaciones (ej. UBE 3 A).
Presenta un s índrome conductual característico con risa inmotivada, aspecto feliz, hiperactividad, hábito de pica, movimientos estereotipados especialmente de manos. Retardo mental, epilepsia con anormalidades electroencefalográficas orientadoras, desorden motor, torpeza motriz y marcha atáxica25-26.
Síndrome de Prader Willi (SPW) (MCK# 176270) (producido en un 70% de los casos por la deleción de la misma región del cromosoma 15q13 de origen paterno, 25% la DUP materna y 2-5% las mutaciones de la zona que regula el imprinting en esa región del genoma.
Estos ni ños suelen mostrarse como severamente hipotónicos en los primeros 18 meses de vida, desarrollando progresivamente un fenotipo conductual reconocido, con apetito voraz con falta de sensación de saciedad y obesidad progresiva secundaria25-27. Suelen mostrar desorden afectivo bipolar, con componentes sicóticos, depresión, ansiedad, o trastorno obsesivo compulsivo. Se han descripto también trastornos del espectro autista con déficit del lenguaje, y de la comunicación e intereses restringidos28.
Estos aspectos no se correlacionan con el grado de compromiso del intelecto, pero s í lo hacen con el mecanismo de producción, ya que aquellos pacientes cuya causa era la DUP o la mutación del centro de imprinting eran más proclives a desarrollar características propias de un TGD.
Otros defectos en esta regi ón (duplicaciones) no vinculables con el SPW o SA dan cuenta de cuadros dismorfológicos diferentes en ocasiones asociados con autismo26 -27.
Deleciones del cromosoma 22q conformando el Síndrome de DiGeorge (McK# 188400), el Síndrome Velocardiofacial (McK #192430) y otras entidades relacionadas, constituyen la deleción intersticial humana más frecuente, abarcando una zona que contiene cerca de 12 genes contiguos29.
Se caracterizan por la asociación de defectos cardíacos cono-truncales, dismorfias faciales y otras malformaciones que comprometen en mayor o menor medida los derivados de las bolsas faríngeas embrionarias.
Pueden presentar inmunodeficiencia, hipocalcemia, paladar hendido o insuficiencia velopalatino, con retardo madurativo global y un fenotipo conductual que puede variar entre el déficit atencional, trastornos del espectro autista, anomalías de la interacción social, desorden del humor, ansiedad y psicosis en la adolescencia y adultez.
Síndrome de Williams (SW) (McK #194050) debido a la deleción de genes contiguos a nivel del cromosoma 7q11.23. Su fenotipo característico con un perfil cognitivo típico es la consecuencia de la haplo-insuficiencia de unos 20 genes, entre los que se encuentra el gen de la elastina30.
Si bien suelen ser hipersociables, no tienen adecuado contacto social con pares, suelen mostrar perseveraciones, estereotipias, ansiedad y berrinches. Si bien no es lo habitual, la asociación entre SW y trastornos del espectro autista ha sido reportada por diversos autores10, 25.
Estas aberraciones cromosómicas estructurales, que pueden estar asociadas a cuadros del espectro autista, fácilmente identificables por la evaluación médico clínica, son muchas veces confirmadas mediante técnicas de FISH con sondas fluorescentes específicas.
Recientemente la implementación de nuevas técnicas citogenéticas como la CGH o Hibridación genómica comparativa mediante microarrays, permite descartar anomalías cromosómicas (duplicaciones o deleciones) de porciones extremadamente pequeñas de los cromosomas (100 Kb), la que suelen escapar a la identificación mediante otros estudios16-17. Estas técnicas prometen ampliar nuestra capacidad de despistaje diagnóstico de anomalías cromosómicas submicroscópicas.
1. B) Monogénicos o mendelianos:
Síndrome de X-frágil (SFX) o de Martin Bell (McK # 300624), Si bien en varones con un fenotipo específico con o sin antecedentes familiares claros este síndrome es fácilmente sospechado, frente a varones o mujeres con trastornos del espectro autista sin causa clara debe descartarse el SFX, ya que el no reconocerlo determinará un inadecuado asesoramiento genético familiar.
Producido por la expansión progresiva de una isla del trinucleótido CGp, ubicada en el extremo 5´ o proximal del gen FRM1.
Dicha isla en la población normal presenta entre 6 y 54 repeticiones, los individuos permutados con 55 a 200 repeticiones y los afectados con mutación completa tienen más de 200 repeticiones al tiempo que la región se encuentra anormalmente metilada, lo que inhibe la trascripción del gen y origina la ausencia de la proteína FRMP31.
El cuadro clínico suele ser claro en varones con retardo mental moderado a severo, mientras que las mujeres pueden presentar un amplio espectro clínico desde la normalidad, trastornos de aprendizaje, trastornos del espectro autista, hasta el RM severo.
La comorbilidad de SFX con TGD se ha estimado entre el 7 y 25%32.
Recientemente Hayashi et al., en un interesante trabajo observaron una mejoría del comportamiento, disminución de estereotipias y ansiedad entre otros aspectos, en ratones knockout FXS al inhibir una kinasa p21 (PAK) la cual tiene un rol crítico en la polimerización de la actina y en la morfogénesis de las espinas dendríticas. Dicha proteína actúa en relación directa con la proteína FMRP33.
Síndrome de Sotos (McK #117550), caracterizado por sobre-crecimiento, conformación cráneo-facial característica, y retardo mental. Suele presentar trastornos en el lenguaje tanto en su iniciación tardía, inadecuada en cantidad y calidad, como trastornos en la articulación de la palabra atribuida a su conformación bucal y posición dentaria34.
El defecto molecular es identificable en un 80-90% de los casos como debido a mutaciones del gen NSD1 (Nuclear receptor SET domain containing protein) (Locus en cromosoma 5q35) (McK *606681)35.
Complejo Esclerosis Tuberosa (McK #191100) debido a mutaciones en el locus génico TSC1 en el cromosoma 9q34 (McK* 605284) y en el TSC2 ubicado en el cromosoma 16q13.3 (McK* 191092)36-37-38.
Caracterizado por la presencia de manchas hipocrómicas, adenomas sebáceos, en muchas ocasiones este complejo se asocia a retardo mental, epilepsia, trastornos de conducta y trastornos del espectro autista.
Los pacientes con mutaciones del gen TSC2 muestran una enfermedad más severa, con mayor frecuencia de convulsiones, incluyendo espasmos infantiles, mayor compromiso cognitivo, retraso madurativo y mental, y mayor ocurrencia de autismo.
El defecto molecular subyacente determina un incremento en los somas neuronales y de las dendritas sinápticas con anomalías en las sinapsis glutamínicas, especialmente en áreas frontales y temporales.
En ambos tipos la asociación con autismo se reporta en un 40-70% de los casos.
En el grupo de pacientes con autismo, esta causa constituye usualmente un 1-4% de los mismos. En nuestra casuística estuvo presente en el 3% de los casos (10).
Otras entidades menos conocidas o frecuentes como el Síndrome de Cornelia de Lange (McK #122470), el Síndrome de Niikawa Kuroki (McK 147920), el Síndrome de Ohdo (McK 249620), y el Síndrome Acrocalloso (McK *606681), pueden ser evidenciables entre los niños con autismo, y su correcta identificación en la consulta genética permitirá orientar los estudios médicos a fin de identificar la entidad en cuestión, posibilitando el asesoramiento de las familias en forma adecuada39.
1. C) Factores ambientales: han sido observados como causantes del espectro autista, generalmente insultos prenatales entre los que deben destacarse las infecciones por: rubéola, CMV y toxoplasmosis, siendo la rubéola una entidad claramente prevenible a través de la vacunación.
Sin duda, el alcohol constituye el grupo más prevalente y más fácilmente erradicable mediante campañas de educación que adviertan a la población en riesgo sobre su potencial efecto teratogénico del comportamiento.
Otros factores como los antiepilépticos (ácido valproico), sustancias abortivas como el Misoprostol, los sucedáneos del ácido retinoico usado en el tratamiento del acné, y los inhalantes como el benceno o el tolueno (ya sea por uso laboral o como droga alucinógena) muestran efectos sobre el desarrollo temprano del tejido nervioso que se traduce luego en trastornos del desarrollo temprano como retardo mental y autismo39.
Grupo 2) Formas no sindrómicas
Este grupo constituye aproximadamente, según diversos autores, el 84% de los pacientes con el espectro autista16-17.
Múltiples estudios han permitido estimar numerosas teorías sobre su etiopatogenia.
Es claro que no responden a un modelo monogénico clásico.
Casi un 25% de ellos muestra asociación con rasgos dismórficos, usualmente menores, no presentes en sus padres sanos, que indicarían efectos ambientales que afectaron el desarrollo embrionario temprano.
Este hallazgo representa un factor predictivo negativo respecto a la respuesta terapéutica, ya que usualmente estos niños responden peor al tratamiento que aquellos en los que no es posible detectar dismorfias evidentes.
La ocurrencia de microcefalia en estos casos, tiene también un valor predictivo de mal pronóstico respecto al tratamiento, no así la macrocefalia que no muestra correlación con la respuesta terapéutica.
A partir de las pruebas moleculares y epidemiológicas, la teoría más aceptada es la del efecto Poligénico: con Heterogeneidad genética (es decir que defectos en distintos genes pueden producir un fenotipo semejante), estimándose que una veintena de genes podrían estar involucrados, y Heterogeneidad alélica (defectos en un mismo gen pueden producir variantes del fenotipo).
Las metodologías utilizadas para evidenciar genes candidatos han arrojado numerosas evidencias aunque resta aún por comprender mucho más.
Distintas regiones del genoma han sido identificadas, como la ubicada en el cromosoma 7q designada AUTS1, donde podrían haber más de 2 genes candidatos.
Otras regiones que muestran evidencias significativas son: 2q, 17q, 15q, 21q entre los más destacados16, 40-42.
Con la intención de facilitar los estudios que permitan identificar genes candidatos, existe fuerte coincidencia en la necesidad de mejorar la selección de las muestras a ser estudiadas, estableciendo criterios más estrictos respecto a las distintas variables fenotípicas tanto físicas como conductuales.
En tal sentido grandes avances se lograron al homogeneizar el muestreo de pacientes de acuerdo a la presencia o no de lenguaje verbal, la edad de emisión de la primer palabra o la primer frase, la presencia de estereotipias, la historia de regresión en las pautas madurativas adquiridas, el sexo de los probandos, el tamaño del perímetro cefálico (normal, macro o microcefalia) entre los más utilizados.
Algunos de estos resultados son expuestos en la Tabla 243-51.
TABLA 2.- Relación entre algunos aspectos fenotípicos y su correlato con hallazgos polimórficos en el genoma.

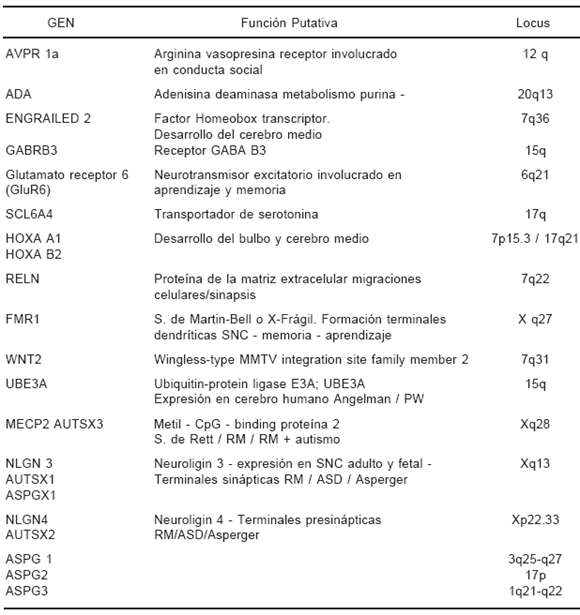
A partir de estos hallazgos y de la búsqueda racional de posibles genes candidatos por el reconocimiento de las funciones que podrían desempeñar en el autismo, se han podido reconocer genes relacionados con el fenotipo en cuestión; algunos de estos hallazgos son resumidos en la tabla Tabla 351.
TABLA 3.- Genes vinculados al autismo51
Fenotipo intermedio: En función de lo antes expuesto es posible comprender la diversidad de fenotipos y la existencia de fenotipos intermedios, producto tal vez de la acción combinada de algunos de los genes causantes del fenotipo completo o más severo.
Estos sujetos tienen cierto compromiso social, de la comunicación, con conductas repetitivas, pero de un modo más sutil por lo que no es posible su encuadre en el espectro autista clásico.
Algunos de los familiares de niños autistas suelen mostrar estas características, habiendo sido reconocido en un 12-25% entre familiares y en un 50% entre padres de probandos52.
Síndrome de Asperger (SA)
Definido como una alteración grave y persistente de la integración social; intereses restringidos y actividades repetitivas según el DSM IV1, con una incidencia estimada en la población general de 2.5/10.0005, este síndrome se diferencia del trastorno autista por la ausencia de retraso significativo en el desarrollo cognitivo y del lenguaje.
El compromiso en la interacción social se manifiesta por compromiso en la comunicación no verbal, contacto ocular, gestos de intención social, incapacidad para establecer relaciones apropiadas con sus pares, hipo-espontaneidad y ausencia de reciprocidad social.
Sus patrones de comportamiento se expresan por intereses restringidos, conductas estereotipadas, obsesiones, preocupaciones absorbentes por un tema, adhesión inflexible a rutinas, estereotipias y preocupaciones por partes de objetos.
En general no hay retraso importante en el desarrollo del lenguaje, incluso éste puede ser extremadamente rico aunque falto de interés comunicativo, lo que puede no ser observado por la familia tempranamente. Tampoco tienen compromiso cognitivo.
Funcionamiento social
A diferencia del síndrome autista los pacientes con SA si bien son socialmente aislados no desconocen la presencia de otros, aunque el acercamiento a sus pares en general no es el apropiado53.
Pueden mantener conversaciones, aunque se interesan en especial en sus propios temas sin dar gran importancia al interés del interlocutor, incluso si éste cambia el tema ellos regresan al de su interés, aunque esto no lleve a ninguna conclusión.
Si bien son solitarios y pueden autodefinirse como tales, son conscientes de sus dificultades para hacer amigos, lo cual en la vida adulta puede llevarlos a trastornos del humor y depresión.
Dada su ausencia de espontaneidad y sus dificultades para comprender emociones e intenciones en forma espontánea o intuitiva; tienen que observar y entender los mismos para responder adecuadamente de manera formal y adecuada, lo que les lleva tiempo y al aprenderlo pueden cumplir con reglas sociales aunque rígidamente
Patrones de comunicación
Si bien el lenguaje oral está comprometido, este es diferente a las formas de autismo más severo.
En el SA se observa un compromiso en la prosodia (especialmente en dar énfasis a las emociones, cambios de humor, etc.).
Pueden tener compromiso en la fluencia e incluso no modular la voz de acuerdo al entorno.
Pueden tener estereotipias verbales, por ejemplo repetir exactamente monólogos de TV
Intereses restringidos
Muchas veces y de acuerdo a su edad pueden tener diversos temas de interés como hacer puzzles; pueden tener habilidades musicales o profundizar en temas como los dinosaurios, la geografía, el universo, etc.
Incluso es aceptado que los temas de interés pueden ir cambiando e incluso dependen del entorno, de la utilidad del mismo, y del abordaje terapéutico53.
Muchas veces los familiares pueden influir en la elección de un tema, por ejemplo uno de nuestros pacientes sabía todas las banderas del mundo, su padre era marino y le había regalado un atlas el cual él miraba y estudiaba compartiendo el reconocimiento con su padre.
Las personas con SA pueden desarrollar conductas agresivas, en especial cuando no se cumplen rutinas establecidas como horarios de comida, itinerarios usuales para llegar a un lugar, etc.
En la adolescencia pueden deprimirse al sentirse aislados e incapaces de tener una interacción social adecuada. Evolución: Si bien las personas con SA no se curan tiene en general una mejor evolución que los autistas de bajo rendimiento, especialmente si sus habilidades especiales les permiten integrarse a un trabajo para su subsistencia independiente; no obstante, estas situaciones son difíciles de lograr.
Aspectos genéticos
Asperger en su descripción original en 1944 ya hizo referencia a características similares en familiares, especialmente en padres.
Esta predisposición familiar fue más recientemente considerada, siendo hoy estimada como la condición psiquiátrica con mayor heredabilidad.
Se ha descrito en asociación a varios síndromes genéticos como síndrome 47, XYY, síndrome de Williams, y síndrome de Sotos, entre otros.
Algunos genes han sido mapeados en autosomas como el ASPG 1: 3q 25-27 (McK 608638), el ASPG 2:17p 13 (McK 608631) y al ASPG 3: en 1q21 - 22 (McK 608631).
En el cromosoma X: ASPGX 1 (McK 300494) por mutación del gen Neuroligin 3 (McK 300336), en Xq13.
Y ASPGX 2 (McK 300497) por mutación del gen Neuroligin 4 (McK 300427) en Xp22.33. (ut supra)54-58.
Trastorno generalizados del desarrollo no especificado (TGD-NOS)
Según el DSM IV1 el TGD-NOS se lo define como una alteración grave y generalizada del desarrollo de la interacción social; de las habilidades de la comunicación verbal y no verbal, o cuando hay intereses restringidos o actividades estereotipadas que no cumplen con el diagnóstico de un trastorno específico del desarrollo o esquizofrenia o trastorno de la personalidad, por lo cual sería un diagnóstico de exclusión más que un patrón diagnóstico específico.
La frecuencia de TGD-NOS en la población general se estima en 15/10.0005.
En realidad si consideramos al síndrome autista como un espectro, podríamos poner al TGD-NOS como una forma más leve diferenciándose del Síndrome Asperger por su mejor nivel cognitivo, su mejor desarrollo del lenguaje y la torpeza motriz presente en el mismo59.
En el caso del autismo muchas veces la diferencia es difícil de evidenciar e incluso, dado que niños autistas al crecer mejoran sus habilidades cognitivas y sociales, en un momento podían ser autistas y al observarlos más adelante entrar en el diagnóstico de TGD - NOS.
La pregunta es, si no conocemos la historia previa, ¿un niño o adolescente con TGD- NOS ha sido siempre así o es un autista que mejoró?
Esta pregunta puede ser difícil de responder aunque lo importante será conocer las fortalezas y debilidades del mismo, en el momento que llega a nuestra consulta, para el abordaje terapéutico y la búsqueda de posibles entidades neurológicas asociadas.
Finalmente luego de haber analizado estos tres subtipos de TGD creemos que el reconocimiento de los diversos subtipos posibles será fundamental para la investigación, ya que diversos patrones, cognitivos y conductuales podrían responder a diversas bases genéticas.
Aspectos neuropsicológicos
A continuación analizaremos algunas de las hipótesis propuestas desde el punto de vista neuropsicológico como implicadas en la génesis del autismo, el síndrome de Asperger y los trastornos generalizados del desarrollo no especificados: Déficit en las funciones ejecutiva, débil coherencia central, trastorno en la cognición social y la teoría de la empatización y sistematización.
Déficit de las funciones ejecutivas (FE)
La conducta humana está organizada por sistemas jerárquicos de respuesta, desde simples reflejos hasta un tope máximo la "Central Ejecutiva" (CE)60.
Shallice en 198861 analiza el funcionamiento de la CE a través del concepto del Sistema de Supervisión Atencional; este sistema selecciona y controla los más bajos niveles de procesamiento automático de rutinas, una función importante en las situaciones nuevas.
Un funcionamiento adecuado de la FE permitirá una adecuada memoria de trabajo y planificación.
Personas con lesiones importantes en la corteza prefrontal tienen compromiso en las funciones ejecutivas, inflexibilidad e incapacidad de planificación.
El compromiso de ciertas FE podrían ser las responsables de las conductas repetitivas, los intereses restringidos y la ausencia de flexibilidad en los individuos autistas.
Débil coherencia central (CC)
Definido como un estilo de procesamiento cognitivo, las personas con una fuerte CC tendrán gran capacidad para observar los aspectos de un todo y extraer lo importante, mientras quienes tengan una débil CC, pondrán gran atención a los detalles sin encontrar lo sustancioso, característica observada en muchos autistas.
Justamente los autistas tendrán mayor capacidad para encontrar figuras escondidas que las personas normales y serán menos susceptibles a las ilusiones ópticas62.
Fink et al.63 en un trabajo realizado con voluntarios normales a los que se le solicitaba que atendiesen a aspectos globales y locales de figuras complejas, observaron que cuando la mirada era dirigida a observación global se activaba el área medial de la misma corteza extra estriada, mientras que cuando la atención era focalizada se activaba el área lateral de la misma63.
Ring et al.64 compararon sujetos normales y autistas a través de Resonancia Magnética Funcional (RMF) a los cuales se les solicitaba que observaran una figura escondida en una imagen y que luego observaran en forma pasiva una pantalla en blanco sin imagen; detectaron que los autistas tenían mayor activación del área lateral de la corteza extra estriada (habitualmente más activada ante la observación de zonas específicas en una figura), mientras que los controles normales tenían mayor activación de la corteza dorso lateral prefrontal64. Estos hallazgos permiten inferir que la capacidad de observación focalizada en autistas está intacta mientras que la capacidad para observación global está comprometida.
Todas estas interesantes observaciones, si bien preliminares, nos permiten evocar el compromiso de la CC en el autismo.
Trastorno en la cognición social
Analizaremos dos aspectos esenciales de la cognición social (CS) que claramente están comprometidos en el autismo: el reconocimiento de caras y la ceguera mental.
Reconocimiento de caras: Nos permitirá no solo reconocernos sino percibir qué es lo que siente o piensa otra persona, interpretar sentimientos. Justamente los autistas tienen serios problemas en diversos aspectos de la percepción de caras y el reconocimiento facial65.
Ceguera mental o ausencia de teoría de la mente: Es la incapacidad de los autistas en comprender conductas de los otros, deseos, inferir segundas intenciones, es considerada fundamental para explicar muchos de los problemas de comunicación e interacción social de los mismos.
La amígdala parece tener un rol importante en el normal desarrollo de la cognición social.
Relacionada con nuestras respuestas emocionales, el reconocimiento del significado afectivo del estímulo, memoria a largo plazo, orientación del estímulo social, percepción de orientación de la mirada, asociaciones cruzadas. La amígdala juega un rol fundamental en el reconocimiento de caras y a través de ellas la percepción de las emociones, en especial la de temor.
Los autistas padecen un compromiso en el reconocimiento de caras, lo que secundariamente redunda en un defecto en la percepción de expresiones faciales y por ende la percepción de las emociones.
El compromiso en la percepción de caras y emociones generaría la imposibilidad de inferir segundas intenciones, comprender miradas, deseos y conductas de los otros, la denominada ceguera mental o ausencia de la teoría de la mente comprometiendo severamente la conducta social.
Justamente las deficiencias que se han relacionado al compromiso de la amígdala son entre otros: Déficit en el reconocimiento facial66, déficit en la detección de expresión de emoción, en especial de miedo67, compromiso en la mirada egocéntrica y pobre lectura de los ojos68.
Recientemente estudios con Resonancia Magnética Funcional (RMNF) en autistas comparados con controles normales pusieron en evidencia la falta de activación de la amígdala derecha en respuesta al estímulo social, en relación a la percepción de la mirada de la cara, en especial de los ojos69 y la falta de activación del girus fusiforme, menor activación de la amígdala y girus occipital, procesamiento de la imagen en áreas diferentes y menor tamaño de la amígdala en autistas en relación a normales70.
Desde el punto de vista neuropatológico, si bien no son muchos los reportes, se hallaron anormalidades consistentes en el sistema límbico, el cerebelo y la oliva Inferior71.
Si bien todo esto no es suficiente para explicar la patogénesis, sabemos que tanto en humanos como en animales las estructuras mediales del lóbulo temporal, en especial el hipocampo y la amígdala tienen importancia en la cognición, el aprendizaje, las emociones y la conducta.
Teoría de la empatización y sistematización (ES)
Baron-Cohen et al. conceptualizan el espectro autista como una mezcla de déficit y fortalezas (las cuales se expresan en forma inadecuada).
Es así que parte de los síntomas del EA podrían explicarse por trastornos en la empatización y excesiva sistematización72.
Empatización: La Empatía, definida como la atribución de los estados mentales de uno mismo y de otros73, es lo que nos permite dar sentido o entender acciones de otras personas, y reaccionar adecuadamente a los estados mentales de los otros.
Incluye la teoría de la mente, la lectura de la mente, el reconocimiento de intenciones y la empatía.
La empatización puede estar comprometida en diversos grados, incluso las personas con Síndrome de Asperger pueden comprender emociones simples pero no complejas
Sistematización: Así como la empatización es deficitaria, algunos aspectos cognitivos de las personas con autismo pueden estar sobre-expresados, lo que podrían determinar parte de sus síntomas, y ser una respuesta para compensar otras deficiencias.
El autista tiende a sistematizar, analizar objetos y eventos para comprenderlos y poder predecir futuras conductas. Como ejemplos de sistemas que están en su entorno y tienden a ser utilizados observamos los técnicos; ej. equipos de música, sistemas abstractos como elementos de computación, matemáticas, geográficos naturales, geológicos, etc.
Analizando estos dos conceptos74 propone que la tríada de déficit relacionada a trastornos en la empatización son el compromiso social, el trastorno en la comunicación y el pobre desarrollo de teoría de la mente, mientras que la hipersistematización estará relacionada a las islas de habilidad, obsesiones por sistemas y conductas repetitivas.
Incluso se puede relacionar la débil coherencia con la teoría de la sistematización.
Si observamos que los autistas sistematizan mejor actividades de pequeñas partes, analizan mejor detalles individuales que el todo (como ejemplo tenemos el reconocimiento de figuras escondidas) e incluso se dirigen mejor a pruebas y ocupaciones que envuelven análisis de sistemas y reglas, es probable que el estilo de CC observado en los autistas sea complementario de la sistematización.
Es así que los autistas de alto rendimiento padecerían una ausencia de los procesos por los cuales las relaciones globales son establecidas.
De lo analizado hasta aquí podríamos conceptualizar el cerebro en dos aspectos 1- el Cerebro social cuyo compromiso se expresa como déficit en la empatía y 2- un Cerebro analítico en el cual una hipersistematización podría llevar a un hiperfuncionamiento analítico.
Esta falta de posibilidad de comprender el todo, haría que el autista sufra la incapacidad de predecir situaciones, llevándolo a analizar una y otra vez una situación para poder entenderla, predecirla y fundamentalmente integrarla a su esquema (hacer las cosas predecibles).
Justamente esto expresaría la necesidad de monotonía para su tranquilidad y predecibilidad y la angustia que le generan los cambios75.
Un correlato neurobiológico posible de la hipersistematización podría ser la hiperconectividad, la cual podría tener una explicación en los hallazgos descriptos por Casanova et al.76 en cerebros de 9 personas autistas fallecidas, en quienes encontraron un número aumentado de minicolumnas con menor número de neuronas en cada una de ellas, siendo las mismas más pequeñas con menor espacio para proyecciones gabaérgicas con la consecuente disminución del GABA.
Estos hallazgos serían coincidentes con las teorías de la inhibición gabaergica77 en la génesis del autismo, relacionadas con el cromosoma 15 (donde se encuentran tres subunidades de genes receptores del GABA) y el cromosoma 3 (en el cual se identificó el gen Gat 1 que sintetiza una proteína que trabaja con el GABA).
Esta anormalidad en las minicolumnas podría generar mayor especificidad con menor generalización y en consecuencia generar una hipersistematización responsable de conductas repetitivas, perseveraciones y una débil coherencia central.
La conectividad entre neuronas es fundamental tanto a nivel focal y como a distancia para desarrollar verdaderas redes neuronales.
Un aspecto interesante a considerar, son las observaciones de trastornos en la empatía y en la sistematización en padres y familiares cercanos de individuos autistas.
Es así que Hugues et al.78 observaron mayor rigidez, ansiedad y tendencia a la soledad en padres de autistas, Baron-Cohen y Hammer en 199779 identificaron un rendimiento sobre lo normal en el reconocimiento de figuras escondidas y Happe et al. 200180 detectaron mayores habilidades en pruebas de alta sistematización.
Por lo que podemos inferir que los padres de autista, tienen una tendencia a padecer alteraciones en la empatía y/o tendencia a la sistematización.
Justamente Baron-Cohen et al. en 199781, analizando ocupaciones de padres y familiares cercanos de autistas observaron una mayor representación de ocupaciones como ingeniería mientras que en Baron-Cohen et al. en 199882 reportaron mayor incidencia de autistas en familiares de ingenieros, matemáticos o físicos.
Estos signos sutiles presentes en familiares cercanos no autistas de autistas pueden ser particularmente informativos en la etiología y en la base genética del autismo, ya que podrían representar diferencias primarias que no fueron enmascaradas por el síndrome autista83, tendencia a la sistematización no sobre-expresada sin tener además compromiso de la empatía.
Finalmente, podríamos definir al autismo como una consecuencia de hipersistematización (con falta de comprensión global) y déficit de la empatización (ausencia de comprensión de estados mentales, teoría de la mente, etc.74).
Condiciones y entidades neurológicas asociadas al autismo
A continuación analizaremos condiciones y entidades neurológicas asociadas al autismo, las cuales es importante conocer ya que nos permitirán realizar un correcto diagnóstico etiológico, un asesoramiento familiar adecuado y un mejor abordaje terapéutico.
Condiciones asociadas
La epilepsia y el retardo mental son dos condiciones neurológicas asociadas al autismo con alta incidencia y son claros indicadores de una disfunción cerebral asociada al mismo.
Epilepsia: Se sabe que entre el 25 y 35% de los autistas padecen epilepsia, con dos claros picos de incidencia según edad, el primero entre los 0 y 3 años y el segundo en la adolescencia84.
En nuestra casuística la incidencia fue del 16 %, este menor porcentaje probablemente esté relacionado con que el promedio de edad de nuestra población fue de 5 años y medio, previo al segundo pico de incidencia10. Retardo Mental: Se estima que entre el 70 y 80% de los autistas padece retardo mental85.
El 95% de nuestros pacientes lo padecieron; el hecho que nuestro estudio se haya desarrollado en un servicio de neuropediatría y no de psiquiatría probablemente sea la explicación para esta mayor incidencia (no obstante en nuestro grupo no se habían incluido deprivados sensoriales ni pacientes con compromiso motor)10.
Tanto la mayor incidencia de Epilepsia como la de Retardo Mental son claros indicadores de una disfunción cerebral asociada al autismo.
Síndrome de Rett
Descrito por Andres Rett en 196686, quizás sea el mejor ejemplo de conductas autistas biológicamente determinadas, donde las afectadas cumplen un patrón conductual y evolutivo sin importar los aspectos sociales, ambientales o familiares.
Esta enfermedad afecta casi exclusivamente a mujeres, con una frecuencia es de 1/10.000 nacidas de sexo femenino87.
El fenotipo clásico es reconocible en más del 75% de los casos, existiendo sin embargo formas atípicas como: variantes congénitas, y formas frustras con preservación del lenguaje, entre otras.
Desde la identificación del gen afectado, MECP2, el cual justifica los síntomas en el 80% de los casos aproximadamente, se han podido reconocer formas atípicas dependientes de diversas mutaciones (según veremos más adelante) incluso varones afectados sin los síntomas típicos de SR, lo que ha justificado la búsqueda de la anormalidad del gen en un espectro mayor de niños autistas sin las formas clásicas de este síndrome88, 89.
Los criterios de SR clásico y sus etapas evolutivas han sido claramente descriptos90.
En principio, este síndrome podrá ser evocado ante toda niña con estancamiento de su crecimiento cefálico, pérdida del uso propositivo de las manos, conductas estereotipadas de sus manos (ej. actitud de lavado, golpeteo y chupeteo) y deterioro psicomotor progresivo.
Biología molecular
En 1999, Amir pudo identificar el defecto molecular subyacente al SR, la mutación del gen MECP2 (McK 300005991.
Este se ubica en el cromosoma Xq28, región sujeta a procesos normales de inactivación genómica.
El MECP2 se transcribe desde el telómero al centrómero del cromosoma X, y contiene 4 exones. La proteína que de él deriva posee 486 aminoácidos, y presenta dos dominios funcionales, el MBD o dominio Methyl- CpG-binding, y el TRD o dominio de represión transcripcional.
MUTACIONES MECP2
Más de la mitad de las mutaciones, que son ciertamente muy numerosas, se producen en alguna de estas dos regiones o en la porción intermedia, siendo usualmente el X paterno el más comúnmente afectado (Figura 192, 93). Esta proteína se une al ADN, más concretamente al dinucleótido CpG, actuando como mediador en la represión de genes.
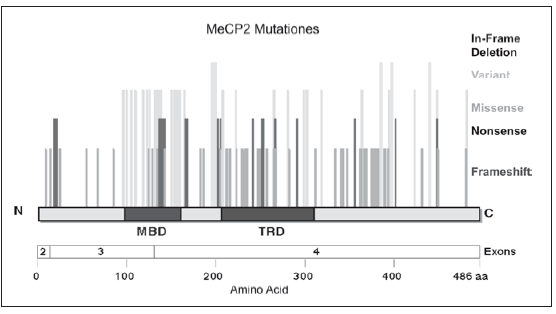
Fig. 1.- Esquema gen MECP2. Dominios MBD y TDR. Tipos de mutaciones descriptas: variables, Missense, Mutación nonsenses y Frameshift92-93.
Algunas de las mutaciones del gen producen proteínas que conservan cierta funcionalidad, lo que permite explicar los casos descritos en varones y otras formas clínicas menos graves del SR e incluso cuadros clínicos de retardo mental, epilepsia y autismo sin las características propias del SR94, 95.
Otros casos en varones son el producto de mosaicismos som áticos del gen, que muestran un cuadro semejante progresivo pero no letal. También se ha descrito SR en personas con cariotipo 47, XXY o varones 46, XX (sexo reverso)96.
Weaving describi ó recientemente formas precoces y atípicas debidas a las mutaciones en un segundo gen posiblemente responsable, el CDKL5 Cyclin-dependent kinase like 5, ubicado en el Xp22. (McK 3000203)97.
La confirmaci ón del diagnóstico en todo paciente en que se sospeche SR es de capital importancia a los fines de determinar su evolución, así como efectuar el consejo genético apropiado. Usualmente se estima que el riesgo de repetición en hermanos es muy bajo, aunque hay descripciones de casos familiares, los que fueron interpretados como consecuencia de otros defectos posiblemente autosómicos que semejan al SR.
En resumen, se interpreta que el SR es la expresi ón más frecuente y dramática de la mutación del gen MECP2, auque existen muchos otros cuadros con retardo mental, epilepsia y autismo consecuencia de mutaciones de este mismo gen sin las características propias de esta entidad, siendo consideradas en la actualidad como un subtipo de afección o MECP2patías.
Se han desarrollado ratones transg énicos con MECP2 truncada, los cuales muestran fenotipo símil al SR.
Ellos son normales hasta la 6ta. semana de vida cuando desarrollan un deterioro neurol ógico progresivo, con compromiso motor, hipoactividad, ansiedad, epilepsia y movimientos estereotipados.
Recientemente, Guy y col. lograron retrasar la aparici ón de los síntomas y revertir el cuadro neurológico en dos pequeños grupos de ratones transgénicos usando una novedosa técnica de ingeniería genética98.
Estos resultados, si bien son muy alentadores, deben a ún ser objeto de más investigación.
Trastorno desintegrativo de la infancia (TDI)
Este raro trastorno es definido en el DSM IV1 como una marcada regresión en múltiples áreas cognitivas y conductuales, entre 2 y 9 años de edad, en un niño previamente sano.
Se define como la pérdida clínicamente significativa de por lo menos dos de las siguientes habilidades adquiridas previamente: Lenguaje (receptivo o expresivo), habilidades sociales, comportamiento adaptativo, control esfinteriano y aparición de conductas del espectro autista.
Este cuadro también denominado Síndrome de Heller99 debe diferenciarse del autismo (trastorno del desarrollo) y de la esquizofrenia infantil.
Dada la rareza de este síndrome, frente a un niño con estas características deben plantearse diversas entidades neurológicas que pueden generar deterioro de este tipo.
Entre ellos podemos citar: 1) Cuadros epilépticos como la afasia epiléptica adquirida o Síndrome de Landau Kleffner (aunque en éste se compromete el lenguaje, sin gran compromiso social), 2) Metabolopatías como la lipofuccinosis infantil ceroidea, la leucodistrofia metacromática, la adrenoleucodistrofia, mitocondriopatías, la gangliosidosis, el Síndrome de Smith Lemli Opitz, etc. 3) Enfermedades infecciosas como la panancefalitis esclerosante subaguda, HIV, entre otras.
Como vemos, frente a un niño con un cuadro de deterioro o pérdida de pautas deben evocarse diversas entidades neurológicas que puedan generar este deterioro, las cuales será importante identificar por sus implicancias evolutivas, terapéuticas y/o genéticas.
Aspectos Genéticos
Si bien las evidencias de una base genética en el TDI son escasas, la misma no debe ser desestimada. Zwaigenbaum reportó dos medios hermanos, uno con autismo y otro con TDI100.
Existe mayor prevalencia de este cuadro en varones que en mujeres con una relación de 4:1, por lo que podría especularse sobre un posible efecto protector del segundo cromosoma X en las mujeres.
No obstante, la ocurrencia de otros TGD en familiares de pacientes con TDI es ciertamente inusual.
Defectos cromosómicos groseros no han sido reportados como posible factor etiológico del mismo.
No se han notificado ocurrencias en hermanos con igual patología por lo que una posible herencia autosómica recesiva podría ser, prima facie, desestimada.
Ante la falta de evidencias en contra, por el momento esta entidad debe ser considerada como esporádica, a la espera de poder determinar su naturaleza neurobiológica y posible riesgo de recurrencia si lo hubiera.
Comentarios finales
Hasta aquí hemos analizado diversos aspectos clínicos, neuropsicológicos y genéticos de los trastornos generalizados del desarrollo; los constantes avances relacionados a la observación conductual, clínica y neurobiológica hacen de este uno de los temas más complejos de la neurología.
La observación conductual, el reconocimiento de fenotipos conductuales autistas, el avance en las investigaciones genéticas, neuropatológicas y neurofisiológicas nos depararán probablemente muchas novedades en los próximos años.
El amplio espectro de presentación clínica, la asociación en muchos casos con entidades neurogenéticas específicas, la importancia de identificación temprana para iniciar un correcto abordaje terapéutico y los constantes avances en los aspectos genéticos hacen que los TGD ameriten un alto grado de compromiso científico y humano por parte de los profesionales que se dedican a su abordaje.
Conflicto de interés: ninguno
Bibliografía
1. Diagnostic and Statistical Manual of Mental Disorders. Am Psychiatric Ass. Washington, DC: DSM IV, 4th ed, 2000 text rev.
2. Kanner L. Autistic disturbances of affective contact. Nev. Child. 1943; 2: 217-250.
3. Asperger H. Die Autistischen Psychopate in kindeslter. Archiv. Fur Pschitrie und nervenkrank heiten 1944; 117: 76-136.
4. Gillberg C. Autism and pervasive developmental disorders. J Child Psychol Psychiat 1990; 31: 99-119.
5. Yeargin-Allsopp M, Rice C, Karapurkar T, Doernberg N, Boyle C, Murphy C. Prevalence of autism in a US metropolitan area. JAMA 2003; 289: 49-55.
6. Miles JH, Hillman R. Value of a clinical morphology examination in autism. Am J. Med.Genet 2000; 91: 245-53.
7. Schopler E, Reichler R, Renner B. The Childhood Autism Rating Scale (CARS) for diagnosis screening and classification of autism. 1986; Irvington, NY: Irvington.
8. Lord C, Rutter M, Le Couteur A. The Autism Diagnosis Interview - Revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Develop Disord 1994; 24: 659-85.
9. Lord C, Rutter M, Goode S. Heemsbergen J, Jordan H, Mawhood et al. Autism Diagnostic Observation Schedule: A standardized observation of communicative and social behaviour. J Autism Develop Disord 1989; 19: 185-212.
10. Ruggieri V, Soprano A, De Carlo A. Aspectos Neurológicos en niños con espectro autista. Medicina Infantil 2005; 12: 192-8.
11. Folstein S, Rutter M. Infantile autism: A genetic study of 21 twin pairs. J Child Psychol Psychiatr 1977; 18: 297-321.
12. Ritvo ER, Jorde LV, Mason Brothers A. The UCLA University of Utah epidemiological survey of autism prevalence. Am J Psychiatry 1989; 146: 194-99.
13. Bailey A, Le Couteur A , Gottesman I, Bolton P, Simonoff E, Yuzda E. et al. Autism as strongly genetic study disorder: Evidence from a British twin study. Psychol Med 1995; 25: 63-78.
14. Jorde LB, Mason-Brothers A, Waldmann R, Ritvo ER, Freeman BJ, Pingree C, et al. The UCLA-University of Utah epidemiologic survey of autism: genealogical analysis of familial aggregation. Am J Med Genet 1990; 36: 85-8.
15. Battaglia A, Carey JC. Etiologic Yield of Autistic Spectrum Disorders. Am J Med Genet 2006; 142 C: 3-7.
16. Muhle R, Treantacoste SV, Rapin I. The Genetics of autism. Pediatrics 2004; 113-5: 472-86.
17. Miles JH, Takahashi TN, Bagby S, Sahota PK, Vaslow DF, Wang CH, et al. Essential versus complex autism: definition of fundamental prognostic subtypes. Am J Med Genet 2005; 135 A: 171-80.
18. Capone GT, Grados MA, Kaufmann WE, Bernad-Ripoll S, Jewell A. Down Syndrome and Comorbid Autism- Spectrum Disorder: Characterization using the aberrant behavior Checklist. Am J Med Genet 2005; 134A: 373-80.
19. Jacobs PA, Price WH, Court Crown WM, Brittain RP, Whatmore PB. Chromosome studies on men in maximum security Hospital. Ann Hum Genet 1968; 31: 339-58.
20. Ruud A, Arnesen P, Stray LL, Vildalen S, Vesterhus P. Stimulant medication in 47,XYY syndrome: a report of two cases. Develop Med Child Neurol 2005; 47: 559-62.
21. Gillberg C. Chromosomal disorders and autism. J Autism Dev Disord 1998; 28: 415-25.
22. Creswell C, Skuse D. Autism in association with Turner syndrome: implication for male vulnerability. Neurocase 2000; 5: 511-8
23. Castelli F, Frith C, Happe F, Frith U. Autism, Asperger syndrome and brian mechanism for the attribution of mental states to animated shapes. Brain 2002; 125: 1839-949
24. Ruggieri V, Arberas C. Autismo en dos niños con Síndrome de Pallister-Killian ¿Configura un fenotipo conductual? Rev Neurol 1999; 29: 261 Abst.
25. Ruggieri VL, Arberas CL. Fenotipos conductuales. Patrones neuropsicológicos biológicamente determinados. Rev. Neurol 2003; 37: 239-53.
26. Borgatti R, Piccinelli P, Passoni D, Raggi E, Ferrarese C. Pervasive developmental disorders and GABAergic system in patients with inverted duplicated chromosome 15. J Child Neurol 2001; 16: 911-4.
27. Dykens EM, Sutcliffe JS, Levitt P. Autism and 15q11-q13 disorders: behavioral, genetic, and pathophysiological issues. Ment Retard Dev Disabil Res Rev 2004; 10: 284-91.
28. Descheemaeker M, Govers V, Vermeules P, Fryns J. Pervasive developmental disorders in Prader-Willi syndrome: the Leuven experience in 59 subjects and controls. Am J Med Genet A 2006; 140 (11): 1136-42.
29. Manning MA, Cassidy SB, Clericuzio C, Cherry AM, Schwartz S, Hudgins L et al. Terminal 22q deletion syndrome: a newly recognized cause of speech and language disability in the autism spectrum. Pediatrics 2004; 114: 451-7.
30. Mervis CB, Morris CA, Bertrand J. Williams syndrome: findings from an integrated program of research. In: Tager-Flusberg H (ed) Neurodevelopmental Disorders 1998. MIT Press, Cambridge, MA, pp 65-110.
31. Goodlin-Jones BL, Tassone F, Gane LW, Hagerman RJ (Autistic spectrum disorder and the fragile X premutation. J Dev Behav Pediatr 2004; 25: 392-8.
32. Cornish K, Kogan C, Turk J, Manly T, James N, Mills A., et al. The emerging fragile X premutation phenotype: evidence from the domain of social cognition. Brain Cogn 2005; 57: 53-60.
33. Hayashi M, Shankaranarayana Rao B, Seo J, et al Inhibition of P21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci USA 2007; 104: 11489-94.
34. Ball L, Sullivan M, Dulany S, Stading K, Schaefer G. Speech-language characteristics of children with Sotos syndrome. Am J Med Genet 2005; 136 A: 363-7.
35. Kurotaki N, Harada N, Shimokawa O, Miyake N, Kawame H, Uetake K, et al. Fifty microdeletions among 112 cases of Sotos syndrome: low copy repeats possibly mediate the common deletion. Hum Mutat 2003; 22: 378-87.
36. Smalley SL, Tanguay PE, Smith M, Gutierrez G (1992) Autism and tuberous sclerosis. J Autism Dev Disord 1992; 22: 339-55.
37. Gillberg IC, Gillberg C, Ahlsen G. Autistic behaviour and attention deficits in tuberous sclerosis: a populationbased study. Dev Med Child Neurol 1994; 36: 50-6.
38. Baker P, Piven J, Sato Y. Autism and tuberous sclerosis complex: prevalence and clinical features. J Autism Dev Disord 1998; 28: 279-85.
39. Cass H, Sekaran, Baird G. Medical investigation of children with autistic spectrum disorders. Child Care Health Dev 2006; 32: 521-33.
40. Bespalova IN and Buxbaum JD. Disease susceptibility genes for autism. Ann Med 2003; 35: 274-81.
41. Wassink TH, Piven J, Vieland VJ, Pietila J, Goedken RJ, Folstein SE, et al. Examination of AVPR1a as an autism susceptibility gene. Mol Psychiatry 2004; 9: 968-72.
42. Veenstra-Vander Weele J and Cook EH Jr Molecular genetics of autism spectrum disorder. Mol Psychiatry 2004; 9: 819-32.
43. Buxbaum JD, Sileverman JM, Smith CJ, Kilifarski M, Reichert J, Hollnder E, et al. Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am J Hum Genet 2001; 68: 1514-20.Epub 2001 May 14. Erratum in: Am J Hum Genet 2001; Aug; 69: 470.
44. Bradford Y, Haines J, Hutchinson H, Gardiner M, Braun T, Sheffield V. et al. Incorporating language phenotypes strengthens evidence of linkage to autism. Am J Med Genet 2001; 105: 539-47. Erratum in: Am J Med Genet 2001 Dec 8; 105 (8): 805.
45. Alarcon M, Cantor R, Liu J, Gilliam T, Geschwind D. Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. Am J Hum Genet 2002; 70: 60-71.
46. Shao Y, Cuccaro M, Hauser E, Raiford K, Menold M, Wolpert C. et al. Fine mapping of autistic disorder to chromosome 15q11-q13 by use of phenotypic subtypes. Am J Hum Genet 2003; 72: 539-48.
47. Buxbaum J, Silevrman J, Keddache M, Smith C, Hollander E, Ramoz N. et al. Linkage analysis for autism in a subset families with obsessive-compulsive behaviors: evidence for an autism susceptibility gene on chromosome 1 and further support for susceptibility genes on chromosome 6 and 19. Mol Psychiatry 2004; 9: 144-50.
48. Molloy C, Keddache M, Martion I. Evidence for linkage on 21q and 7q in a subset of autism characterized by developmental regression. Mol Psychiatry 2005; 10: 741-6.
49. Stone J, Marriman B, Cantor R, Yonan A, Gillian T, Geschwind D. et al. Evidence for sex-specific risk alleles in autism spectrum disorder. Am J Hum Genet 2004; 75: 1117-23.
50. Cantor R, Kono N, Duvall J, Alvarez-Retuerto A, Stone J, Alarcon M. et al. Association testing in a linked region using large pedigrees. Am J Hum Genet 2005; 76: 538-42.
51. Bacchelli E, Maestrini E. Autism Spectrum Disorders: Molecular genetic Advances. Am J Med Genet 2006; 142 C: 13-23.
52. Bishop D, Maybery M, Wong D, Hallmayer. Characteristics of the broader phenotype in autism: a study of siblings using the children's communication checklist-2. Am J Med Genet B Neuropsychiatr Genet 2006; 141: 117-22.
53. Klin A, Mcpartland J, Volkmar F. Asperger Syndrome in Handbook of autism and pervasive developmental disorder. Edit. Vlolkmar F, Rhea P, Klin A, Cohen D. Hoboken, New Jersey John Wiley & Sons Inc. 2005; 3rd edition 88-125.
54. Auranen, M, Vanhala, R, Varilo, T, Ayers, K, Kempas, E, Ylisaukko-oja. et al. T. A genomewide screen for autism spectrum disorders: evidence for a major susceptibility locus on chromosome 3q25-27. Am J Hum Genet 2002; 71: 777-90.
55. Tentler D, Johannesson T, Johansson M, Rastam M, Gillberg C, Orsmark C, et al. A candidate region for Asperger syndrome defined by two 17p breakpoints. Europ J Hum Genet 2002; 11: 189-95.
56. Ylisaukko-oja T, Nieminen-von Wendt T, Kempas E, Sarenius S, Varilo T, von Wendt L, et al. Genome-wide scan for loci of Asperger syndrome. Molec Psychiat 2004; 9: 161-8.
57. Rehnstrom K, Ylisaukko-oja T, Nieminen-von Wendt T, Sarenius S, Kallman, T, Kempas E. et al. Independent replication and initial fine mapping of 3p21-24 in Asperger syndrome. (Letter) J Med Genet 2006; 43: e6.
58. Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg I, et al. Paris Autism Research International Sibpair Study: Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nature Genet 2003; 34: 27-9.
59. Towbin K. Pervasive developmental disorder not otherwise specified. In Handbook of autism and pervasive developmental disorder. Edit. Volkmar F, Rhea P, Klin A, Cohen D. Hoboken, New Jersey John Wiley & Sons Inc. 2005; 3rd edition 155-200.
60. Baddeley AD. Working memory. Oxford Clarendon Press 1986.
61. Shallice T. From neuropsychology to mental structure 1988; Cambridge University Press, Cambridge.
62. Happé F. Autism: cognitive deficit or cognitive style? Trends Cogn Sci 1999; 3: 216-22.
63. Fink GR, Halligan PW, Marshall JC, Frith CD, Frackowiak RSJ, Dolan RJ. Neural mechanisms involved in the processing of global and local aspects of hirarchically organized visual stimuli. Brain 1997; 120: 1779-91.
64. Ring HA, Baron-Cohen S, Wheelwright S. et al. Cerebral correlates of preserved cognitive skills in autism-a functional MRI study of embedded figures tasks performance. Brain 1999; 122: 1305-15.
65. Blair RJR, Frith U, Smith N, Abell F, Cipolloti N. Fractionation of visual memory: agency detection and its impairment in autism. Neuropsychologia 2002; 40: 108-18.
66. Dawson G, Carver L, Meltzoff AN, Pangioitides H, McPartland J, Webb SJ. Neural correlates of face and object recognition in young children with autism spectrum disorder, developmental delay, and typical development. Child Dev 2002; 73: 700-17.
67. Howard MA, Cowell PE, Bowcher J, et al. Convergent neuroanatomical and behavioural evidence of an amygdala hypothesis of autism. Neuroreport 2000; 11: 2931-5.
68. Calder AJ, Lawrence AD, Keane J. Reading the mind from the eye gaze. Neuropsychologia 2002; 40: 1129-38.
69. Baron-Cohen S, Ring HA, Wheelwright S, Bullmore ET, Williams SE. Social intelligence in the normal and autistic brain: an fMRI. Eur J Neurosci 1999; 11: 1891-8.
70. Pierce, Muller RA, Ambrose J, Allen G, Courchesne E. Face processing occurs outside the fusiforme "face area" in autism: evidence from functional MRI. Brain 2001; 124: 2059-73.
71. Bauman ML, Kemper TL. Hystoanatomic observations of the brain in early infantile autism. Neurology 1985; 35: 866-74.
72. Baron-Cohen S. The extreme male brain theory of autism. Trends Cogn Sci 2002; 6: 248-54.
73. Baron-Cohen S. Mindblindness: An essay on autism and Theory of Mind. 1995 Cambridge, MA MIT Press.
74. Baron-Cohen S, Belmonte M. Autism: A window onto the development of the social and the analytic brain. Annu Rev Neurosci 2005; 28: 109-26.
75. Baron-Cohen S. The cognitive neuroscience of autism. J Neurol Neurosurg Psychiatry 2004; 75: 945-8.
76. Casanova M, Buxhoeveden D, Switala A, Roy E. Minicolumnar pathology in autism. Neurology 2002; 58: 428-32.
77. Hussman P. Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J Autism Dev Disord 2001; 2: 247-8.
78. Hughes C, Leboyer M, Bouvard M. Executive function in parents of children with autism. Psychol Med 1997; 27: 209-20.
79. Baron-Cohen S, Hammer J. Parents of children with Asperger syndrome: What? is the cognitive phenotype? J Gogn Neurosci 1997; 9: 548-54.
80. Happe F, Briskman J, Frith U. Exploring the cognitive phenotype of autism: weak "central coherence" in parents and siblings of children with autism: I. Experimental tests. J Child Psychol Psychiatry 2001; 42: 299-307.
81. Baron-Cohen S, Wheelwright S, Stott C, Goodyer L. Is there a link between engineering and autism? Autism 1997; 1: 153-63.
82. Baron-Cohen S, Bolton P, Wheelwright S, Scahill V, Short L. Autism occurs more often in families of physicists, engineers, and mathematicians. Autism 1998; 2: 296-301.
83. Belmonte MK, Cook EH Jr, Anderson GM, Rubenstein JL, Greenough WT, Beckel-Mitchener A, et al. Autism as a disorder of neural information processing: directions for research and targets for therapy. Mol Psychiatry 2004 9: 646-63.
84. Volkmar F, Nelson DS. (1990) Seizure disorders in autism. Am Acad Adolesc Psychiaty 1990; 29: 127-9.
85. Fombonne E. The epidemiology of autism: a review. Psychol Med 1999; 29: 769-86.
86. Rett A. Ueber ein eigenartiges hirnatrophisches Syndrom bei Hyperammoniamie in Kindesalter. Wien Med Wschr 1966; 116: 723-38.
87. Rett Syndrome Diagnostic Criteria Work Group. Diagnostic criteria for Rett syndrome. Ann Neurol 1988; 23: 425-8.
88. Zappella M, Gillberg C, Ehlers S. The preserved speech variant: a subgroup of the Rett complex: a clinical report of 30 cases. J Autism Dev Disord 1998; 28: 519.
89. Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, et al. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann Neurol 2000; 47: 670-9.
90. Hagberg B, Hanefeld F, Percy A, Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome: comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol 2002; 6: 293-7.
91. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat Genet 1999; 23: 185-8.
92. Hoffbuhr K, Devaney JM, LaFleur B, Sirianni N, Scacheri C, Giron J et al. MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology 2001; 56: 1486-95.
93. Charman T, Neilson TC, Mash V, Archer H, Gardiner MT, Knudsen GP et al. Dimensional phenotypic analysis and functional categorisation of mutations reveal novel genotype-phenotype associations in Rett syndrome. Eur J Hum Genet 2005; 13: 1121-30.
94. Burford B. Perturbations in the development of infants with Rett disorder and the implications for early diagnosis. Brain Dev 2005; 27: S3-7.
95. Glaze DG. Neurophysiology of Rett syndrome. J Child Neurol 2005; 20: 740-6.
96. Kankirawatana P, Leonard H, Ellaway C, Scurlock J, Mansour A, Makris CM. et al. Early progressive encephalopathy in boys and MECP2 mutations. Neurology 2006; 67: 164-6.
97. Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OLD, Archer H, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet 2004; 75: 1079-93.
98. Guy J, Gun J, Selfridge J, Cobb S, Bird A. Science 2007; 315: 1143-7.
99. Heller T. Dementia Infantilis Zeitschrift fur die Erforschung und Behandlung des Jugenlichen Schwachsinns 1908; 2: 141-65.
100. Zwaigenbaum L, Szatmari P, Mahoney W, Bryson S, Bartolucci G, MacLean J. High functioning autism and childhood disintegrative disorder in half brothers. J of autism and Dev Disorders 2000; 30: 121-6.

