










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957
Acta bioquím. clín. latinoam. v.43 n.4 La Plata oct./dic. 2009
SECCIÓN PERMANENTE LATINOAMERICANA
Anomalías morfológicas de los leucocitos en el niño
O. Fenneteau1
1. Service d'Hématologie Biologique, Hôpital Robert Debré, Paris, AP-HP.
Editor: Dr. Camilo Fernández Espina
Colaboración: Dr. Miguel Blasco Vega
Asociación Española de Farmacéuticos Analistas
Modesto La fuente, 3 - 28010 Madrid
AEFA agradece a Biologiste et Praticien las facilidades y autorización desinteresada para la traducción al español y la inserción de sus artículos en Laboratorio y Clínica. Los autores de los originales no son, en ningún caso, responsables de la absoluta fidelidad en la traducción de los mismos.
Resumen
Se realiza una revisión de las anomalías morfológicas de polinucleares, monocitos y linfocitos precisando su orientación diagnóstica en el niño. En efecto, es al principio de la vida cuando se manifiestan la mayoría de las patologías hereditarias que tienen repercusión en los leucocitos.
PLAN
1. Anomalías morfológicas de los polinucleares
2. Anomalías morfológicas de los monocitos
3. Anomalías morfológicas de los linfocitos
Los leucocitos, células sanguíneas circulantes de la serie blanca, pueden presentar diferentes anomalías morfológicas en los frotis sanguíneos coloreados con May Grünwald Giemsa (MGG). El reconocimiento de estas anomalías permite orientar el diagnóstico de diferentes patologías, constitucionales o no, que son las que serán abordadas.
Serán tratadas sucesivamente las anomalías citoplasmáticas (granulaciones anormales, vacuolas) y nucleares en cada categoría celular (polinuclear, monocito, linfocito).
1. Anomalías morfológicas de los polinucleares
1.1. INFECCIONES SEVERAS
En el curso de las infecciones bacterianas severas, cualquiera que sea la edad, los polinucleares neutrófilos pueden presentar diversas anomalías citoplasmáticas asociadas: granulaciones tóxicas, intensificación de las granulaciones o, al contrario, desgranulación, presencia de cuerpos de Döhle (zona basófila), vacuolas, así como anomalías nucleares: defecto de segmentación o hiper-segmentación, núcleos dobles, gigantismo. Gérmenes y también parásitos, levaduras, pueden ser observados en los polinucleares neutrófilos (Figura 1).
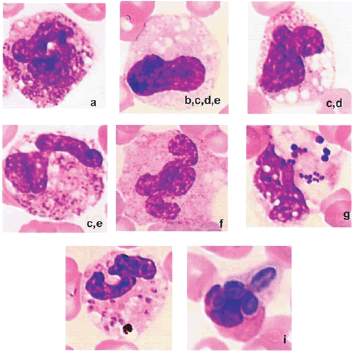
Figura 1. En el curso de las infecciones bacterianas severas, cualquiera sea la edad, los polinucleares neutrófilos presentan anomalías citoplasmáticas diversamente asociadas: granulaciones tóxicas, fortalecimiento de las granulaciones, o al contrario, desgranulación (b), presencia de cuerpos de Döhle (espacio basófilo) (c), de vacuolas (d), así como anomalías nucleares (déficit de segmentación (e) o hiper-segmentación, núcleos dobles (f), gigantismo). Gérmenes (g), y también parásitos (merozoitos de Plasmodium falciparum) (h), levaduras (i) pueden ser observadas en los polinucleares neutrófilos.
1.2. MIELODISPLASIAS
La mielodisplasia es rara en el niño (1); las anomalías morfológicas de los polinucleares son idénticas a las observadas en el adulto: desgranulación o granulación anormal, a veces bastoncillos de Auer, defecto de segmentación, doble núcleo, núcleo en forma de cinta (Figura 2).

Figura 2. Las mielodisplasias en el niño son escasas. Las anomalías morfológicas de los polinucleares son idénticas a las observadas en el adulto: desgranulación (a) o granulación anormal (b), a veces bastoncillos de Auer (c), defecto de segmentación (d), dobles núcleos, núcleo en cinta (f).
1.3. CONDENSACIÓN ANORMAL DE LA CROMATINA
En algunos pacientes con trasplantes (de órganos, de médula ósea) bajo tratamiento inmunosupresor, se pueden descubrir anomalías de la estructura de la cromatina en el frotis sanguíneo. Los polinucleares neutrófilos tienen un núcleo poco lobulado con una cromatina fragmentada y condensada (Figura 3). Este fenómeno retrocede después de suspender el tratamiento (2).

Figura 3. En ciertos pacientes injertados (trasplantados de órganos, de médula ósea), bajo tratamiento inmunosupresor, se pueden hallar en el frotis sanguíneo anomalías de la estructura de la cromatina. Los polinucleares neutrófilos tienen un núcleo poco lobulado con cromatina fragmentada y condensada.
1.4. ENFERMEDADES HEREDITARIAS
1.4.1. Anomalías citoplasmáticas
En el curso de la enfermedad de Chediak-Higashi, enfermedad hereditaria de transmisión autosómica recesiva, que asocia albinismo oculocutáneo parcial, infecciones severas y un déficit inmunitario, las granulaciones de todos los polinucleares son anormales (Figura 4). Son dispersas, ensanchadas, de coloraciones diversas con MGG. Los lisosomas de secreción son anormales (3). La granulación de los eosinófilos está aumentada, con aspecto cristaloide.

Figura 4. En el curso de la enfermedad de Chediak-Higashi, enfermedad hereditaria, las granulaciones de todos los polinucleares son anormales (a). Son dispersas, ensanchadas, de coloraciones diversas en el MGG. Las granulaciones de los eosinófilos son de gran tamaño y aspecto cristaloide (b).

En el curso rhohhde las enfermedades de sobrecarga lisosomal, los polinucleares neutrófilos pueden presentar granulaciones anormales, testigos de sobrecarga lisosomal, esencialmente en el curso de mucopolisacaridosis (MPS) (4). Así, la anomalía de Alder se traduce en la presencia de granulaciones azurófilas más groseras, más numerosas, anormalmente cargadas de mucopolisacáridos (Figura 5).

Figura 5. En el curso del mucopolisacaridosis la anomalía de Alder se traduce por la presencia de granulaciones azurófilas más groseras, más numerosas, anormalmente cargadas de mucopolisacáridos. La anomalía es localizable en los polinucleares eosinófilos, con granulaciones de tinte variable (grises, rojo oscuro, verduscas).
El aspecto puede ser parecido a las granulaciones tóxicas observadas en los polinucleares neutrófilos en infecciones severas, pero sin otras anomalías citológicas (vacuolas, cuerpos de Döhle). La anomalía es localizable más fácilmente en los polinucleares eosinófilos con granulaciones de tinte variable (gris, roja oscura, verdusca). Se observan siempre en la enfermedad de Maroteaux-Lamy (MPS de tipo VI), pero pueden ser inconstantes en el curso de otras MPS.
El aspecto de los polinucleares neutrófilos de los pacientes afectados por MPS de tipo IV (enfermedad de Morquio) es particular, con granulaciones raras pero grandes, a veces reagrupadas en pares (Figura 6).

Figura 6. El aspecto de los polinucleares neutrófilos de los pacientes afectados por MPS de tipo IV (enfermedad de Morquio) es particular, con granulaciones escasas pero de gran tamaño, a veces agrupadas en pares.
La presencia de polinucleares eosinófilos anormales (Figura 7) se observa en pacientes que sufren sialidosis y gangliosidosis GM1 en asociación con linfocitos con vacuolas múltiples.

Figura 7. La presencia de polinucleares eosinófilos anormales se observa en pacientes que sufren sialidosis y gangliosidosis GM1 (a) en asociación con linfocitos con vacuolas múltiples. En el curso de mucosulfatidosis, las granulaciones están dispersas (b).
En el curso de una mucosulfatidosis, las granulaciones eosin ófilas aparecen dispersas.
Tales im ágenes de gránulos, poco numerosos, se encuentran también en pacientes afectados por el raro déficit hereditario en peroxidasa de los eosinófilos, cuyo núcleo a menudo está hipersegmentado.
En las hipereosinofilias reactivas (alergias, tratamiento por factor de crecimiento) pueden observarse im ágenes similares (Figura 8).
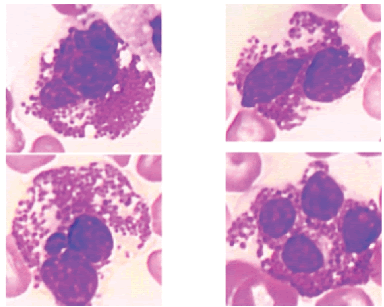
Figura 8. Hipereosinoflia reactiva, granulaciones dispersas y plurisegmentación son posibles.
En el curso de diversas enfermedades metabólicas, se pueden observar vacuolas citoplasmáticas lipídicas coloreadas en rojo por Oil Red O (ORO) (anomalía de Jordans), como el síndrome de Dorfman-Chanarin (5), el déficit en beta-oxidación de ácidos grasos (Figura 9) y el déficit de carnitina.

Figura 9. En el curso de enfermedades diversas y metabólicas, se pueden observar vacuolas citoplasmáticas lipídicas coloreadas en rojo por Oil Red O (ORO) (anomalía de Jordans). Ejemplo de déficit en acil-CoA deshidrogenasa de ácidos grasos de cadena mediana (MCAD).
En el curso del síndrome MYH9 (6) es evocadora la asociación de inclusiones basófilas en los polinucleares y trombopenia con plaquetas de tamaño aumentado (Figura 10).

Figura 10. En el curso del síndrome MYH9, es sugerente la asociación de inclusiones basófilas en los polinucleares y de trombopenia con plaquetas de gran tamaño. La anomalía de May-Hegglin se caracteriza por la presencia de una o a veces dos voluminosas inclusiones basófilas (a), mientras que en los síndromes de Fechtner y de Sebastián (b), las inclusiones son más discretas.
La anomal ía de May-Hegglin se caracteriza por la presencia de una o, a veces 2, inclusiones voluminosas basófilas (pseudocuerpos de Döhle), mientras que en el síndrome de Fechtner y de Sebastián, las inclusiones son más numerosas y más discretas y evidenciar su presencia requiere un examen atento. Estas inclusiones se deben a la precipitación en el citoplasma de miosina no muscular de tipo IIA, anormal por mutación del gen MYH9.
1.4.2. Anomalías del núcleo
Las anomalías se refieren al grado de segmentación nuclear o a la textura de la cromatina.
La anomalía de Pelger-Huet, sin repercusión funcional, corresponde a una hiposegmentación del núcleo en dos lóbulos.
En la hipersegmentación constitucional de Undritz se observan más de cinco lóbulos en la mayoría de los polinucleares.
En el curso de los trastornos hereditarios de la vitamina B12 o de los folatos, los polinucleares neutrófilos pueden ser hipersegmentados o más frecuentemente encintados (en banda) (Figura 11).

Figura 11. En el curso de los trastornos hereditarios de vitamina B12 o de los folatos, los polinucleares neutrófilos pueden estar hiper-segmentados (a) o más frecuentemente en cintas (b).
En el curso de mielocatexis como en el síndrome de WHIM (7), la morfología de los polinucleares neutrófilos es muy particular y asocia vacuolas citoplasmáticas a un núcleo con varios lóbulos entrelazados por filamentos largos. Este aspecto se observa sobre todo en la médula ósea donde los polinucleares son numerosos (Figura 12) mientras que en sangre periférica, existe neutropenia.

Figura 12. En el curso de mielocatexis, como en el síndrome de WHIM, la morfología de los polinucleares neutrófilos es muy particular y asocia vacuolas citoplasmáticas a un núcleo de varios lóbulos conectados entre ellos por filamentos largos. Este aspecto se observa sobre todo en la médula ósea en la que los polinucleares son numerosos, mientras que la sangre periférica es neutropénica.
En el curso de la intolerancia a las prote ínas dibásicas (8), enfermedad metabólica rara, a menudo de revelación neonatal, que se acompaña de anemia y de trombopenia, se observan en la sangre polinucleares neutrófilos picnóticos.
En la m édula ósea es muy evocadora la presencia de polinucleares picnóticos, cuyo núcleo se reduce a una masa redondeada con fagocitosis selectiva de estos núcleos por histiocitos y precursores mieloides (Figura 13).

Figura 13. En el curso de la intolerancia a las proteínas dibásicas, enfermedad metabólica rara de frecuente revelación neonatal, que se acompaña de anemia y trombopenia, se observan en sangre polinucleares neutrófilos picnóticos (a). En la médula ósea es muy sugerente la presencia de polinucleares picnóticos, cuyo núcleo se reduce a una masa redondeada con fagocitosis selectiva de estos núcleos por histiocitos (b) y precursores mieloides (c).
Se observan anomal ías en el curso de la trisomía 13 y de la trisomía 14 con mosaicismo, la mayoría de los polinucleares neutrófilos presentan en el núcleo proyecciones sésiles o pedunculadas (Figura 14).

Figura 14. En el curso de trisomía 13 y trisomía 14 con mosaicismo, la mayoría de los polinucleares neutrófilos presenta en el núcleo proyecciones sésiles o pedunculadas
2. Anomalías morfológicas de los monocitos
2.1. INFECCIONES SEVERAS
Los monocitos son de gran tamaño, fuertemente vacuolados, con una cromatina más fina.
2.2. LEUCEMIA MIELOMONOCITARIA JUVENIL
Un fuerte monocitosis, asociada en niños pequeños (<4 años) a esplenomegalia voluminosa es evocadora de esta patología. Los monocitos a menudo tienen un aspecto dismórfico con fuertes repliegues y cromatina más fina. El frotis sanguíneo también revela la presencia de eritromielemia y escaso número de blastos mieloides (Figura 15).

Figura 15. La leucemia mielomonocitaria juvenil asocia, en los niños pequeños, esplenomegalia voluminosa a una fuerte monocitosis. Los monocitos a menudo tienen un aspecto dismórfico con grandes repliegues, cromatina más fina (a). El frotis sanguíneo también revela la presencia de eritromielemia (b) y blastos mieloides en bajo número (c).
2.3. ENFERMEDADES HEREDITARIAS
2.3.1. Anomalías citoplasmáticas
En el curso de la enfermedad de Chediak-Higashi, se observan a veces en el frotis sanguíneo monocitos raros con una inclusión de tinte variable rosa pálido a rojo (Figura 16).

Figura 16. En el curso de la enfermedad de Chediak-Higashi, a veces se observan en el frotis sanguíneo escasos monocitos con una inclusión de tinte variable, de rosa pálido a rojo.
En el curso de las enfermedades de sobrecarga lisosomal se encuentran granulaciones voluminosas de tinte morado oscuro en los monocitos de los pacientes afectados por mucopolisacaridosis, particularmente de tipo I, VI, VII (Figura 17).

Figura 17. En el curso de las enfermedades de sobrecarga lisosomal, las granulaciones voluminosas de tinte morado oscuro se encuentran en los monocitos de los pacientes afectados de mucopolisacaridosis, particularmente de tipo I, VI, VII.
En el curso de la enfermedad de May-Hegglin, pueden observarse zonas bas ófilas en los monocitos.
2.3.2. Anomalías nucleares
En el curso de enfermedades metabólicas raras, particularmente la aciduria metilmalónica, asociada a un déficit de metimalonil-coenzima A mutasa, se puede observar cromatina desestructurada en los monocitos y en menor grado en los polinucleares neutrófilos (Figura 18).

Figura 18. En el curso de enfermedades metabólicas raras, particularmente en la aciduria metilmalónica, asociada a un déficit de metimalonilcoenzima A mutasa, se puede observar cromatina desestructurada en los monocitos (a), y en menor grado, en los polinucleares neutrófilos (b).
Tales imágenes de cromatina "deshecha" también se encuentran en patologías adquiridas, particularmente en el curso de grandes hipertermias
3. Anomalías morfológicas de los linfocitos
3.1. INFECCIONES
En el curso de la tos ferina (Tos convulsa) que puede sobrevenir en el lactante, es casi invariable la hiperlinfocitosis de linfocitos maduros que presentan, no siempre, un aspecto particular con núcleo hendido (Figura 19). En el curso del síndrome mononucleósico son numerosas las células mononucleadas hiperbasófilas. Su aspecto es heterogéneo en tamaño, intensidad de basofilía y en textura de la cromatina. Las células más típicas tienen 15-20 µm de diámetro, pueden presentar un fortalecimiento de la basofilia en periferia, un núcleo a menudo en forma de bandera (Figura 20) y granulaciones finas azurófilas.

Figura 19. En el curso de la tos ferina en lactantes, es casi constante la hiperlinfocitosis de linfocitos maduros, de los que todo un contingente presenta un aspecto particular con un núcleo hendido, aunque este aspecto no es constante.

Figura 20. En el curso del síndrome mononucleósico, están en gran número las células mononucleadas hiperbasófilas. Su aspecto es heterogéneo en tamaño, en intensidad de basofilia y en la textura de la cromatina. Las células más típicas tienen 15-20 µm de diámetro, un fortalecimiento de la basofilia en periferia, un núcleo a menudo en bandera.
3.2. ENFERMEDADES HEREDITARIAS
Las anomalías morfológicas de los linfocitos en el curso de las enfermedades metabólicas hereditarias únicamente afectan al citoplasma, pero son diversas.
En el curso de la enfermedad de Chediak-Higashi, la acumulación de proteínas lisosomales en lisosomas de secreciones afecta a los linfocitos T citotóxicos y a las células natural killer (NK), lo que se traduce por la presencia de una, o más raramente, varias granulaciones voluminosas rojo vivo al ser coloreadas con May Grünwald Giemsa (Figura 21).

Figura 21. En el curso de la enfermedad de Chediak-Higashi, la acumulación de proteínas lisosomales en los lisosomas de secreciones está presente en Iinfocitos T citotóxicos y células natural killer (NK) y se traduce en la presencia de una o más granulaciones voluminosas de color rojo vivo al May Grünwald Giemsa (MGG).
En el curso de la mucopolisacaridosis (9), el hallazgo de linfocitos de Gasser es un elemento importante de orientación diagnóstica (Figura 22). El linfocito de Gasser se caracteriza por la presencia de una o, a veces varias, granulaciones gruesas de tinte violeta oscuro, más raramente rosadas con May Grünwald Giemsa, en el seno de una vacuola. Linfocitos de Gasser también se pueden hallar en el curso de la enfermedad de Austin (mucosulfatidosis) y a veces en el curso de gangliosidosis de GM2.

Figura 22. En el curso de la mucopolisacaridosis (MPS), el hallazgo de linfocitos de Gasser es un elemento importante de orientación diagnóstica. El linfocito de Gasser se caracteriza por la presencia de una, o a veces varias, granulaciones grandes de tinte morado oscuro, más raramente rosado al MGG, en una vacuola. Linfocitos de Gasser también se pueden encontrar en el curso de la enfermedad de Austin (mucosulfatidosis) y a veces en el curso de gangliosidosis GM2.
Linfocitos con granulaciones anormales también son testigos de una sobrecarga de MPS. En uno o dos polos del linfocito, en general de pequeño tamaño, se concentran granulaciones anormales más o menos oscuras, a veces dentro de una vacuola voluminosa. Estas granulaciones presentan un metacromasia al azul de toluidina (Figura 23). Este tipo de linfocito es frecuentemente observado en la MPS de tipo III (Enfermedad de Sanfilippo).

Figura 23. Linfocitos con granulaciones anormales también testimonian sobrecarga MPS. En uno o dos polos del linfocito, en general de pequeño tamaño, se concentran granulaciones anormales más o menos oscuras a veces en el seno de una vacuola voluminosa. Estas granulaciones presentan metacromasia al azul de toluidina (b).
Los linfocitos de Gasser y los linfocitos con granulaciones anormales no deben ser confundidos con linfocitos granulados: linfocitos T citotóxicos y células natural killer (Figura 24).

Figura 24. Los linfocitos de Gasser y los linfocitos con granulaciones anormales no deben ser confundidos con linfocitos granulados: linfocitos T citotóxicos y células natural killer.
En el curso de diversas enfermedades de sobrecarga lisosomal (Tabla I), la detección de los linfocitos vacuolados es un elemento de orientación diagnóstica.
Tabla I. Linfocitos y enfermedades de sobrecarga

Es la anomalía citológica más frecuente en las enfermedades de sobrecarga, pero no es específica.
Los linfocitos normales pueden contener microvacuolas o más raramente vacuolas grandes. En las enfermedades de sobrecarga lisosomal, el porcentaje de linfocitos vacuolados es variable, pudiendo alcanzar el 80% o al contrario, ser un porcentaje muy bajo.
Las vacuolas no se colorean por el MGG y por lo tanto, son ópticamente vacías, salvo en la mucolipidosis de tipo II donde algunas vacuolas se colorean de rosa. La presencia de una sobrelínea de vacuolas color rosa (linfocitos rhodocircés) (Figura 25) se observa frecuentemente en ciertas MPS (Enfermedad de Hunter).

Figura 25. Las vacuolas no se colorean con MGG y, por tanto, son ópticamente vacías, salvo en la mucolipidosis de tipo II en las que algunas vacuolas están coloreadas en rosa (a). La presencia de una supralínea vacuolar en color rosa en los linfocitos (linfocitos rhodocircés) (b) frecuentemente se observa en alguna MPS (Enfermedad de Hunter).
Ya sean las vacuolas de pequeño tamaño y escasas o de talla importante y numerosas, en estos casos se habla de "vacuolas múltiples y grandes" (Figura 26). El tamaño y el número de las vacuolas, así como el porcentaje de células afectadas dependen de la enfermedad de sobrecarga en cuestión.

Figura 26. Tamaño de las vacuolas pequeño y en escaso número (a), o grandes y en gran número (b). Se habla, entonces, de "vacuolas múltiples y grandes". El tamaño y el número de las vacuolas, así como el porcentaje de células afectadas dependen de la enfermedad de sobrecarga en cuestión (Cf. Tabla I).
Por ejemplo, la enfermedad de Wolman se puede sospechar ante el hallazgo de linfocitos con vacuolas, de peque ño tamaño, reagrupados en racimos, visibles en el citoplasma o en el núcleo de coloración Oil Red O positiva. Contrariamente, los linfocitos vacuolados en el curso de la enfermedad de Niemann Pick que presenta, en sangre un aspecto semejante, son Oil Red O negativo. En el mielograma en estas dos patologías se observa la presencia de numerosos histiocitos vacuolados (Figura 27). En el curso de la enfermedad de Pompe (Tabla I), las vacuolas de pequeño tamaño son PAS positivas.

Figura 27. Enfermedad de Wolman: presencia de linfocitos con vacuolas, de pequeño tamaño, agrupadas en racimo, visibles en el citoplasma o en el núcleo (de coloración Oil Red O positivo) (a), presencia de numerosos histiocitos vacuolados (b) en la médula ósea.
Puede citarse la gangliosidosis GM1 en la que los linfocitos tienen vacuolas grandes, múltiples, muy visibles, asociadas con polinucleares eosinófilos anormales (Figura 28).

Figura 28. En la gangliosidosis GM1 los linfocitos con vacuolas grandes, múltiples, son muy visibles y están asociados a polinucleares eosinófilos anormales (a).
La búsqueda de elementos a favor de una enfermedad de sobrecarga debe ser objeto de una observación minuciosa de por lo menos 100 linfocitos y de polinucleares neutrófilos. Estas anomalías no son constantes, de manera que en algunos pacientes afectados por una auténtica enfermedad de sobrecarga, pueden no hallarse las anomalías morfológicas encontradas entre otros pacientes (particularmente en la mucopolisacaridosis de tipo III).
La orientación diagnóstica se basa en la apreciación del número y de la asociación eventual de las diferentes anomalías encontradas (Tabla II).
Tabla II. Tipos de sobrecarga lisomal y anomalías citológicas sanguíneas

Todas las anomalías citológicas o nucleares de los leucocitos, cuando están presentes, son una ayuda muy valiosa para la orientación diagnóstica y permiten la práctica rápida de otras pruebas que completen el perfil etiológico.



AGRADECIMIENTOS
El autor agradece a H. Ogier (Hôpital Robert Debré - AP-HP) por sus críticas y su valiosa ayuda y a S. Piveteau (Hôpital Robert Debré - AP-HP) por su excelente trabajo técnico.
1. Hasle H, Niemeyer CM, Chessells JM, Baumann I, Bennett JM, Kerndrup G. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 2003; 17: 277-82. [ Links ]
2. Daliphard S, Accard F, Delattre C, Toupance O, Guyot C, Mechinaud F, et al. Reversible abnormal chromatin clumping in granulocytes from six transplant patients treated with mycaphenolate mofetil: a rare adverse affect mimicking abnormal chromatin clumping syndrome. Br J Haematol 2002; 116: 726-8. [ Links ]
3. De Saint Basile G. Implication du trafic intracellulaire dans trois maladies héréditaires du système hématopolétique. Med Sc (Paris) 2000; 16: 745-50. [ Links ]
4. Smith H. Anomalies of leucocyte structure. Diagnosis in pediatric haematology. New York: Churchill Livingstone 1996. p. 201-38. [ Links ]
5. Srebrnik A, Tur E, Perluk C, Elman M, Messer G, Ilie T, et al. Dorfman-Chanarin syndrome. J Am Acad Dermatol 1987: 17 (S PE 1): 801-8. [ Links ]
6. Pujol-Moix N, Kelley MJ, Hernandez A, Muniz-Diaz E, Español I. Ultrastructural analysis of granulocyte inclusions in genetically confirmed MYH9-related disorders. Haematologica 2004; 89: 330-7. [ Links ]
7. Balabanian K, Lagane B, Pablos JL, Laurent L, Planchenault T, Verola O, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood 2005; 105: 2449-57. [ Links ]
8. Doireau Y, Fenneteau O, Duval M, Perelman S, Vilmer E, Tomati G, et al. Intolérance aux protéines dibasiques avec lysinurie: aspect caractéristique de l'atteinte médullaire. Arch Pediatr 1996; 3: 877-80. [ Links ]
9. Cosson A, Maroteaux P, Tapon J, Maier M, Girot R. Apport de la cytologie sanguine et médullaire au diagnostic des maladies de surcharge. Nauv Rev Fr Hematol 1977; 18: 205-6. [ Links ]

