










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de dermatología
versión On-line ISSN 1851-300X
Rev. argent. dermatol. v.88 n.2 Ciudad Autónoma de Buenos Aires abr./jun. 2007
Diagnósticos diferenciales de la histiocitosis a células de Langerhans
The differential diagnostics of Langerhans cell histiocytosis
C. N. Chirino, R. J. Scwartz , O. L. Musitani
Médico Dermatólogo. Universidad Nacional de Buenos Aires. Profesor Asociado Cátedra de Anatomía. Universidad Católica de Cuyo. Jefe de Sección Dermatología"Policlínico Regional J. D. Perón". Villa Mercedes. San Luis. E-mail: canechi@01hotmail.com.ar
Profesor Titular de la Cátedra de Salud Pública. Universidad Católica de Cuyo. Profesor Adjunto de la Cátedra de Neonatología Patológica. Universidad Católica de Cuyo. Cirujano Infantil, por concurso, P. R. "J. D. Perón". Villa Mercedes. San Luis.
Médica Comunitaria del Programa de Médicos Comunitarios de la Nación. Villa Mercedes. San Luis. Médica Generalista. Ex Residente en Medicina General del Hospital "Olga Stucky de Rizzi". Reconquista. Provincia de Santa Fe.
RESUMEN: La histiocitosis a células de Langerhans (HCL) debe diferenciarse de las siguientes entidades: eritema tóxico neonatorum (ETN), dermatitis seborreica (DS), foliculitis pustulosa eosinofílica (FPE), incontinencia pigmenti (IP), mastocitosis/urticaria pigmentosa (M/UP), acrodermatitis enteropática (ADE), síndrome de Wiskott-Aldrich (WAS), acropustulosis infantil (API). Además se deben considerar la enfermedad de Rosai- Dorfman (ERD), xantomas diseminados, melanosis pustulosa neonatal (MPN), candidiasis congénita, listeriosis neonatal, herpes simple perinatal y la varicela neonatal. Debido a que los métodos auxiliares de laboratorio no siempre están disponibles o los resultados laboratoriales algunas veces son extemporáneos, y puesto que el médico práctico a menudo necesita tomar decisiones precozmente, es que la epidemiología resulta útil, pues brinda el marco adecuado para ordenar y jerarquizar las sospechas diagnósticas frente a un caso concreto, con un paciente determinado, en un momento específico.
PALABRAS CLAVE: Epidemiología; Histiocitosis; Células de Langerhans.
SUMMARY: The differential diagnostics of Langerhans cell histiocytosis should include the following disorders: erythema toxicum neonatorum, seborrheic dermatitis, eosinophilic pustular folliculitis, incontinentia pigmenti, mastocytosis / urticaria pigmentosa, acrodermatitis enteropathica, Wiskott-Aldrich syndrome, infantile acropustulosis, Rosai- Dorfman disease, xanthoma disseminatum, neonatal pustular melanosis, congenital candidiasis, perinatal listeriosis, perinatal herpes simplex, neonatal varicella. Since the auxiliary methods of lab are not always available, or lab results are sometimes extemporaneous, the physicians often needs to make quick decisions. The epidemiology is useful because it offers the appropriate mark to prioritize the diagnostic in specific cases.
KEY WORDS: Epidemiology; Histiocytosis; Langerhans cell.
Fecha de recepción: 09.04.07
Fecha de aceptación: 20.06.07
INTRODUCCIÓN
Este grupo de desórdenes idiopáticos caracterizados por la proliferación de las células de Langerhans tiene numerosos diagnósticos diferenciales. En el presente trabajo se destacan los aspectos epidemiológicos, útiles para efectuar razonamientos durante la consulta efectuada por un paciente determinado.Hemos encontrado el obstáculo del subregistro o peor, la carencia de registro, por eso aquí los datos tomados en cuenta se refieren a cifras de Estados Unidos de Norteamérica (USA) o Europa.
Ante la sospecha de histiocitosis de células de Langerhans (HCL) el médico se pregunta:¿Qué otras entidades son semejantes? Y puesto que la dermatitis seborreica (DS) es más prevalente y tiene mayor incidencia ¿es probable que se trate de esta entidad y no de aquella otra, más severa y menos frecuente? Es conocido que las pruebas diagnósticas disponibles hoy día son perfectibles, como lo han demostrado el análisis epidemiológico y los cálculos estadísticos, por ello actualmente la tendencia es intentar "identificar el problema de salud, tratar de resolverlo y hacerse cargo de los cuidados del enfermo", muchas veces más rentable para el paciente, además de ser el objetivo presente de la tarea médica.
DESARROLLO
La dermatitis seborreica (DS) es un desorden pápuloescamoso localizado en las áreas seborreicas del cuero cabelludo, cara y tronco. Se la ha relacionado con la Malassezia, con anormalidades inmunológicas y con la activación del complemento. El cuadro clínico varía desde la pitiriasis leve a formas eritrodérmicas exfoliativas. Aunque puede estar presente en los neonatos, la DS infantil o del lactante es muy prevalente entre la segunda semana y el sexto mes de vida, con otro pico de incidencia entre la tercera y octava semana. Se ha afirmado que hasta el 70% de los niños padecen pitiriasis, una forma clínica de DS.
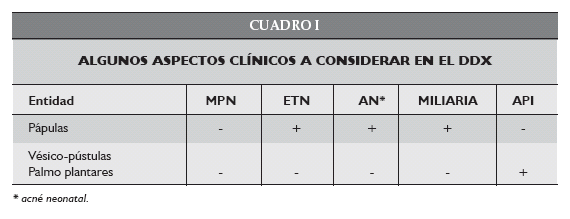
Considerándosela más común que la psoriasis, afecta entre el 2 al 5% de la población, si bien la incidencia exacta no se conoce, se sabe que se incrementa en la pubertad y la clínica más severa en los pacientes HIV positivos (el 85% de ellos tiene DS) y en los afectados por la enfermedad de Parkinson, otra entidad que suele asociar formas más importantes de DS 1. El eritema tóxico neonatorum (ETN) tiene una incidencia en recién nacidos a término del 30-50% y suele debutar a los 4 días de vida (picos de 2 a 14 días). Este cuadro benigno de la piel es asintomático y autolimitado, las lesiones elementales observadas son: pápulas eritematosas pequeñas, vesículas estériles y ocasionalmente, pústulas. Estos elementos están generalmente rodeados por un halo eritematoso difuso distintivo, de aspecto turbio.
Un cuadro que guarda alguna similitud la pustulosis estéril eosinofílica (PEE) en la cual la lesión elemental no está restringida sólo al folículo piloso. Es un desorden recidivante de la piel de etiología desconocida. Observable en niños de 5-10 meses de vida, es excepcional en neonatos. Tiene también picos de incidencia en la 2ª a 4ª década de la vida, pero puede aparecer a cualquier edad; el 10% de la población HIV (+) la padece, en éstos la clínica se torna severa y persistente. Los pacientes con PEE desarrollan múltiples ciclos de exacerbaciones y remisiones, durante estos brotes periódicos aparecen pústulas estériles y pápulas que en el 50% de los casos se acompaña de prurito. Las lesiones generalmente son autoresolutivas en un tiempo variable, entre algunos meses y varios años.
La incontinencia pigmenti (IP) o síndrome de Bloch-Sulzberger (SBS), en su etapa vesicular evocaría posiblemente a la DS, aunque la IP además exhibe pústulas, ampollas y eritema, con disposición Blaschko lineal. Es observable al nacer, pero hay niños mayores con lesiones reactivadas por ciertas enfermedades febriles. La incidencia es baja de 1: 40.000 habitantes. Al ser una genodermatosis HAD-X debe priorizarse su sospecha diagnóstica ante una niña de raza blanca (la proporción es 37 : 1 ).
Este desorden de la pigmentación de la piel, asocia a menudo alteraciones oculares, dentales, y del sistema nervioso central (SNC). La evolución del cuadro implica pérdida de melanina de las células basales epidérmicas. Al principio fue descripta como esporádica y ligada a la banda Xp11.21, luego se supo que estas mutaciones en realidad provocan la hipomelanosis de Ito, o IP tipo 1; el Consenso Internacional de la IP en el año 2000 informó que la IP o SBS es causada por alteraciones de un gen denominado NEMO -o Modulador esencial del factor nuclear kappa B (gamma IKBKG-IKK)- localizado en el cromosoma Xq28, cercano al gen para el factor VIII, las mutaciones del gen generan proteínas truncas.
Dos tercios de estas nuevas mutaciones provienen del padre 2. La mastocitosis / urticaria pigmentosa (M/UP) se caracteriza por la proliferación y acumulación de mastocitos en la intimidad de distintos órganos, siendo el más habitual la piel. Las formas clínicas cutáneas de mastocitosis incluyen el mastocitoma solitario, la mastocitosis eritrodérmica difusa, la mastocitosis paucicelular (o telangiectasia macularis eruptiva perstans TMEP), y la urticaria pigmentosa (UP). La mastocitosis sistémica conlleva acumulación de clones celulares derivadas del mastocito en diversos tejidos, además de la piel; así la médula ósea, el tracto gastrointestinal, el hígado y el bazo pueden estar implicados. La UP es la forma clínica más usual y se caracteriza por máculas, o placas. Aquellas son redondeadas u ovales, de color marrón-rojizo, su cantidad varía en número, de entre algunas a miles. Eventualmente, en la infancia, las lesiones pueden ser vesiculosas. Es referido en USA que el 0,1 al 0,8% de las consultas dermatológicas corresponde a una forma de M/UP; en el Reino Unido es extremadamente rara, la prevalencia registrada es de 2: 300.000 habitantes, la proporción hombre/mujer es igual, y la mayoría de los pacientes son de raza blanca. El 55% corresponde a menores de 2 años, el 10 % entre los 2 a 15 años, con un segundo pico de inicio a los 30-49 años. La UP en los niños suele ser autoresolutiva, aunque se ha observado shock por degranulación masiva. Cuando se inicia en la adolescencia o en la adultez se detecta un aumento del riesgo relativo (RR) de progresión hacia la cronicidad y a las formas sistémicas y del riesgo atribuible (RA) de malignidad, el cual en adultos asciende al 30% y en adolescentes al 7% 3.

La acrodermatitis enteropática (ADE) se produce por un error congénito del metabolismo del zinc, de carácter autosómico recesivo (HAR). Su clínica en la infancia incluye dermatitis periorificial (oral, anal, genital) y acral, diarrea, trastornos neurológicos, infecciones secundarias bacterianas o fúngicas, cambios mentales y de la conducta. Las lesiones eritematoescamosas, eccematosas, erosivas, pustulosas, vesico-ampollares, y su distribución pueden remedar la DS, sin embargo la epidemiología indica que la forma heredada es muy rara, así en Dinamarca, la prevalencia es de 1: 500.000 habitantes.
La dermatitis acral y periorificial también puede observarse en enfermedades no genéticas: 1- en la deficiencia de biotina, 2- en la candidiasis local y generalizada, 3- en la epidermólisis ampollar atípica, 4- en la dermatitis atópica, 5- en las anomalías del metabolismo de los ácidos grasos esenciales, 6- en la dermatitis seborreica, 7- en el déficit de zinc provocado por ingesta inadecuada, mala absorción, pérdida excesiva, o combinación de estos factores. 8- en el kwashiorkor, que históricamente ha sido confundido con formas infantiles de la pelagra, 9- en la pelagra provocada por déficit tisular de niacina (vitamina B3 o ácido nicotínico), y/o de triptofano, un aminoácido esencial precursor de la niacina.


La pelagra es frecuente en dietas abundantes en maíz ya que este es pobre en triptofano, o en dietas con mijo, rico en leucina, pues este aminoácido interfiere en el metabolismo del triptofano. Esta enfermedad es típica de adultos, rara en la infancia, pero observable en jóvenes adolescentes con dietas pelagrogénicas. En nuestro país es desconocida la incidencia y prevalencia de esta avitaminosis, sin embargo, en la población de alcohólicos crónicos se presumen cifras altas. Otras enfermedades asociadas con déficit de niacina son: a- patologías intestinales de niños, b- en la "desnutrición o hambre oculta" de los consumidores de comida chatarra, muy prevalente, rica en grasas y azúcares, pero pobre en micro nutrientes (vitaminas y minerales) c- tratamientos con isoniazida, que compite con el metabolismo de la niacina, del síndrome carcinoide, que utiliza triptofano alternativamente para formar serotonina 4,5,6.
Las HCL son un grupo de desórdenes idiopáticos caracterizados por la proliferación de las células de Langerhans, una especie celular especializada de la médula ósea, y de eosinófilos maduros. En las HCL se han documentado anormalidades en el número y la función de las células supresoras creándose así un sistema de immunovigilancia permisivo, responsable aunque sea en parte, de la patogénesis de estos cuadros.
El grupo de trabajo denominado "Sociedad del Histiocito" ha dividido las alteraciones de estas células en 3 grupos diferentes:
(I) histiocitosis de células dendríticas,
(II) desórdenes del macrófago eritrofagocítico, e
(III) histiocitosis malignas.
La HCL pertenece al grupo I ofreciendo un espectro clínico variable: desde a-una enfermedad aguda fulminante,la enfermedad de Letterer-Siwe (ELS), hasta b- una enfermedad localizada, con lesiones únicas (o escasas), indolentes, crónicas, del hueso u otros órganos (granulomas eosinófilos), c- una forma intermedia, el síndrome de Hand-Schüller-Christian (SHSC), multifocal, crónica, y generalmente con la tríada típica: diabetes insípida, proptosis y lesiones líticas del hueso, d- una forma congénita autolimitada, la enfermedad de Hashimoto-Pritzker (EHP), y e- una forma de histiocitosis cutánea pura.
La patogénesis de HCL es desconocida. Se discute si este proceso es reactivo (A) o neoplásico (B). Argumentos a favor de (A): a- posibilidad de remisiones espontáneas, b- no se han detectado aneuploides (células con un número anormal de cromosomas), c-anormalidades cariotípicas o de metafase, d- alta tasa de sobrevida en pacientes con formas clínicas sin disfunción orgánica.
Argumentos a favor de (B): a- células aberrantes que infiltran órganos, b-posibilidad de evolución mortal, c- tratamiento exitoso con antineoplásicos en ciertas variantes clínicas de HCL, d- existencia de proliferación monoclonal ligada X.
En USA las HCL son raras. La incidencia anual estimada es de 0.5-5.4 casos por millón de habitantes por año. Se informan en ese país casi 1200 casos nuevos anuales. Más del 50% de los niños menores de 2 años, aquejados con formas diseminadas y que cursan con disfunción orgánica, mueren de esta enfermedad. Las formas unifocales y la mayoría de los casos congénitos son autoresolutivas, salvo aquellos lactantes con HCL y compromiso pulmonar, en quienes se ha observado aumento de la mortalidad. Existe mayor prevalencia entre blancos y la proporción varón: mujer es de 2: 1, afectando desde neonatos a adultos. La edad de comienzo varía según la forma clínica: ELS predomina en niños menores de 2 años. La forma multifocal crónica, incluido SHSC tiene un pico de comienzo en niños de entre 2-10 años. Los granulomas eosinofílicos localizados aparecen más frecuentemente entre los 5-15 años de edad 7.
El síndrome de Wiscott-Aldrich (WAS) es un desorden genético recesivo ligado al X, afecta casi exclusivamente a varones, no obstante hay informes en niñas, uno de ellos se refiere a una paciente de 8 años de edad quien presentaba una mutación del gen denominado WASp en el cromosoma X paterno, asociada con inactivación no-al azar de su cromosoma X materno. WAS es una severa inmunodeficiencia congénita, los pacientes afectados a menudo no sobreviven la niñez; aunque en 2 grandes series de casos se informaron enfermos en su cuarta década de vida 8.
El gen defectuoso (WASp), relativamente raro, reside en Xp11.22-23 cuya mutación genera una proteína anómala con expresión limitada a las células de la progenie hematopoyética. En los linfocitos regula un componente del citoesqueleto, la actina, actora, parcialmente, de la morfología, movimiento y crecimiento linfocítico. Sus mutaciones alteran entonces el desarrollo, migración y motilidad no sólo de los linfocitos, sino también de otras células hematopoyéticas (plaquetas, células dendríticas y neutrófilos) responsable así de los inmunodefectos 9.
WAS está caracterizado por 1- hemorragias (el 84% tiene trombocitopenia, pero sólo el 20% tiene anomalías hematológicas), 2-infecciones piógenas recidivantes (sólo el 5% las tiene) y 3-dermatitis recalcitrante. Pero esta tríada clásica sólo se ve en el 27% de los pacientes. Si bien el síndrome es de expresión variable, incluye frecuentemente deficiencias de la IgM y casi siempre (84%), trombocitopenia persistente; en un estudio de 154 pacientes, sólo el 30% presentaba la expresión completa que incluye atopia (eccema), inmunodeficiencia celular y humoral, plaquetas pequeñas, aumento del riesgo relativo de enfermedad autoinmune y malignidades hematológicas.
La incidencia en USA es de 4 casos por millón de varones nacidos vivos. En Suiza es de 4.1 casos por millón de varones nacidos vivos. La prevalencia en Italia, Japón, Suiza, Suecia, Irlanda de WAS, en la población de pacientes con inmunodeficiencias primarias, es del 2-8.8%. Respecto a la morbilidad/mortalidad se ha demostrado en un estudio reciente con pacientes sobrevivientes de entre 1-35 años (media de 11 años), que el tiempo medio de sobrevida fue de 8 años. Las causas de muerte más importante fueron las hemorragias o las infecciones y en una serie, el 12% de los pacientes desarrolló además, malignidades, principalmente tumores linforreticulares o leucemia. En esta serie el RR de malignidades fue mayor a 100. En otro estudio similar las causas de muerte informadas fueron: infecciones (44%), hemorragias (23%), o malignidades (26%). La esplenectomía permite mayor sobrevida y el transplante de médula puede ser curativo. En un grupo de 301 pacientes con WAS, confirmado o probable, provenientes de 149 familias, 8 familias eran negros, y 4 chicanos. De 40 familias cuyo linaje fue remontado fuera de Norteamérica, 38 habían emigrado de Europa. La acropustulosis infantil (API) es una erupción recurrente, auto-limitada, pruriginosa, vesicopustular palmo-plantar que afecta a niños de 2 a 3 años de vida y que probablemente sea mucho más frecuente de lo sugerido por la escasa cantidad de informes publicados.
La patofisiología es desconocida. Muchos casos de API son precedidos por escabiosis, documentada o sospechada, insinuando que aquella es una reacción secundaria a ésta, sin embargo la API no relacionada con sarna es más frecuente. La inmunofluorescencia y los cultivos bacterianos y virales son negativos, por eso se cree que la API no es un proceso autoinmune mediado por anticuerpos. La incidencia exacta es desconocida tanto en USA como internacionalmente. Un estudio en Israel divulgó 25 casos en un período de ocho años, sugiriendo que no es tan infrecuente. Los primeros informes propusieron un predominio en afro-americanos, pero ahora, se cree que afecta a todas las razas por igual, estos informes también hicieron sospechar predominio masculino, pero series más grandes han demostrado una distribución igual entre los varones y mujeres.
La API comienza típicamente en los primeros 2 a 12 meses de vida y resuelve generalmente a los 3 años de edad, a pesar de esto se han descripto casos en niños mayores de 9 años.
Otros diagnósticos diferenciales a considerar son:
1) La enfermedad de Rosai-Dorfman (ERD) o histiocitosis sinusal con linfadenopatía masiva. Es el desorden proliferativo infrecuente de los histiocitos, con un curso a menudo prolongado; el 80% de los pacientes tiene una edad comprendida entre los 10 y 20 años de vida. Es muy rara, los casos publicados rondan los 1.000 casos. La enfermedad tiene una distribución universal con predilección por los negros de África y las Indias Occidentales. Suele presentar fiebre y linfadenopatía no dolorosa extensa, frecuentemente cervical, anemia, neutrofilia, VSG elevada, e hipergammaglobulinemia policlonal.
El 43 % tiene compromiso extraganglionar, la piel es el sitio más comúnmente afectado (10 %); en el 3 % de los casos el compromiso es sólo cutáneo. Las lesiones son pápulas o nódulos castaño-amarillentos, máculas eritematosas, paniculitis o pápulas acneiformes. El 13% tiene también desórdenes inmunes (AC antieritrocitarios, compromiso articular). La etiología aún es desconocida, pero hay numerosos informes identificando al virus humano del herpes 6 (HHV-6) en lesiones viscerales y cutáneas, pero al ser éste un virus ubicuo (muchos desórdenes reactivos e infecciosos del tejido linfoide lo tienen), su presencia en ERD no es específica. Generalmente la evolución es autoresolutiva. Los infiltrados histiocíticos se tiñen positivamente para S100, CD1ç, CD14, CD68, laminina 5, y lisozima 10,11.
2) Xantomas diseminados: Son lesiones caracterizadas por la acumulación de macrófagos colmados de lípidos, resultado de un trastorno celular local o reflejo de alteraciones en el metabolismo lipídico.
Los xantomas son altamente prevalentes y afectan a ambos sexos por igual, a cualquier edad. La mayoría de las veces son sólo un problema estético, pero su presencia debe inducir investigaciones del metabolismo lipídico pues esta dermatosis puede preceder el diagnóstico de hiperlipidemia. Los xantelasmas suelen aparecer en mayores de 50 años y una historia familiar de éstos puede sugerir una hiperlipoproteinemia (HLP) hereditaria, algunos de estos síndromes revelan historias previas de formas de arteriosclerosis, incluida enfermedad coronaria, infartos de miocardio, y/o pancreatitis, situaciones relacionadas con su morbi-mortalidad.
Los lípidos, al ser insolubles en agua, deben ser transportados como apoproteínas específicas o como complejos lipoproteicos que sirven también como ligandos a receptores específicos, facilitando los transportes transmembrana y la regulación de la actividad enzimática.
Hay mutaciones genéticas que producen apolipoproteínas defectuosas (hiper-lipoproteinemias primarias), o desórdenes sistémicos con elevación de los niveles lipídicos (hiperlipoproteinemias secundarias).
Fredrickson, tradicionalmente, clasificó a las hiperlipidemias en 6 fenotipos, según el tipo de lipoproteínas elevadas. Recientemente se ha comprendido mejor la base genética y bioquímica de estos desórdenes revelándose que a pesar de ser clínicamente similares, hay más variantes.
Los xantomas eruptivos, que debutan en la niñez, y asocian triglicéridos elevados y colesterolemia normal, cursan con defectos de la lipoproteínlipasa tisular; es denominado HLP tipo I. Si el defecto es de la apolipoproteína C-II la HLP es llamada tipo V. Estos dos cuadros pueden presentar también pancreatitis. Los xantomas, cuando aparecen en adultos acompañados de lipidograma normal, sugieren HLP tipo IV.
Ateroesclerosis severa acompañada de xantomas tendinosos, xantomas tuberosos y xantelasmas, de debut en la adultez, con triglicéridos normales y colesterol muy elevado hacen pensar en la HLP tipo IIa, provocada por déficit del receptor LDL o por la apolipoproteína B-100 defectuosa. La HLP tipo IIb o combinada, es clínicamente idéntica a la anterior pero presenta trigliceridemia y colesterolemia elevadas. Ateroesclerosis precoz, asociada a enfermedad coronaria, y xantomas, preferentemente planos y palmares, se observan en adultos con mutaciones de las apolipoproteínas E, A-I y C-III, denominada HLP III.
Causas de hiperlipidemia secundaria son: embarazo, hipotiroidismo, colestasis y porfiria aguda intermitente. Causas de hipertrigliceridemia secundaria son el uso de contraceptivos orales, enfermedad por almacenamiento de glicógeno tipo I, diabetes mellitus, sepsis a gram (-), alcoholismo, pancreatitis y gota. Causas de hipercolesterolemia e hipertrigliceridemia secundarias simultáneas, son síndrome nefrótico, el fallo renal crónico, y la corticoterapia12.
3) Melanosis pustulosa neonatal (MPN) Se trata de una dermatosis benigna descripta por Ramamurthy en 1976, debuta en el momento del parto, afecta al 5% de los neonatos de raza negra y a menos del 0,3% de los niños de raza blanca. Presenta vesículas, pústulas superficiales que suelen resolver en 48 horas, y máculas pigmentadas de color castaño que pueden persistir varios meses. Localizadas preferentemente en el mentón, cuello, frente, pecho, y espalda, y más raro en palmas y plantas. No presenta signos sistémicos. Es más frecuente en los nacidos a término, afectando tanto a varones como a niñas.
La etiología es desconocida y no existe relación con infección materna o fetal. No se ha identificado predisposición familiar. El diagnóstico diferencial es con eritema tóxico neonatal, impétigo estafilocócico y otras infecciones bacterianas, sífilis, candidiasis neonatal, infección por herpes virus simple o varicela-zóster, miliaria, foliculitis pustulosa eosinofílica, acropustulosis de la infancia, milium, acné neonatorum, manchas mongólicas 13,14,15,16 (Ver Cuadro I).
4) Candidiasis congénita Se trata de una infección raramente informada (70 casos durante los años noventa en USA), puede ser adquirida por el neonato inútero o durante el parto. Probablemente, la candidiasis congénita es una infección intrauterina ascendente con manifestaciones cutáneas o sistémicas típicamente presentes dentro de las 12 horas después del nacimiento. La forma sistémica congénita es típicamente fatal, aunque usualmente las infecciones cutáneas congénitas tienen un curso más benigno. Los nacimientos a pretérmino y la presencia de un dispositivo intrauterino (DIU) está asociado con esta condición. Sin tratamiento, los neonatos tienen un riesgo superior de desarrollar infección sistémica asociada con una tasa de mortalidad alta (70%). Algunos neonatos tienen distrés respiratorio útero de líquido amniótico contaminado. Las lesiones cutáneas consisten en pápulas eritematosas, eritema difuso y típicamente vesículo-pústulas que al romperse dejan ver el collarete epidérmico. El compromiso pude involucrar toda la piel y mucosas 17,18.
5) Listeriosis neonatal El agente causal es el bacilo Gram (+) Listeria monocytogenes, patógeno intracelular facultativo que al ser de baja virulencia afecta a huéspedes con inmunidad deficiente (especialmente celular): embarazadas, fetos, neonatos, ancianos inmunodeprimidos por medicamentos o enfermedades crónicas. Además de trastornos gastrointestinales, puede provocar sepsis, meningitis, corioamnionitis y feto muerto. Puede originar epidemias y casos esporádicos. Se supone que su transmisión se produciría a través de alimentos contaminados con materia fecal de reservorios animales (ganado bovino, porcino, ovino, aves silvestres), humanos o ambientales, incluido el suelo. Cuando se presenta dentro de la primera semana de vida se denomina listeriosis temprana; después de los siete días: listeriosis tardía. La proporción estadística entre ellas es 2/3 - 1/3 respectivamente. La modalidad temprana o granulomatosis infanto-séptica es de mayor mortalidad (30% a 50%), está asociada con síndrome febril materno, corioamnionitis y parto prematuro, estaría provocada por la aspiración de líquido amniótico infectado. Sus manifestaciones clínicas más frecuentes son sepsis, síndrome de distrés respiratorio, meningitis, alteraciones de la termorregulación, pústulas y petequias en tronco y extremidades.
La modalidad tardía se asocia con parto a término no complicado. Su patogenia es incierta, aunque se considera por transmisión nosocomial (materiales contaminados), y contagio a partir de "portadoras sanas", pacientes con infecciones asintomáticas de la faringe y/o tracto digestivo inferior o de la vía genital. Esta última podría provocar infección ascendente del saco amniótico y del feto, ya sea secundaria a rotura prematura de membranas, o bien por la infección del feto durante su descenso por el canal de parto. La tasa de portadores sanos (3% - 11%) contrasta con la baja incidencia; en USA la listeriosis invasiva confirmada por hemocultivo o cultivo del líquido cefalorraquídeo (LCR) es de 7,4: millón habitantes por año. La listeriosis perinatal complica 16 de cada 100.000 nacimientos y el 23% causan feto muerto. Constituyen el 2% de las causas de muertes fetales en general. En Chile entre los años 1982 y 1987 la incidencia fue de 0,42 por 1.000 recién nacidos vivos, con una letalidad de 17,6%. En Argentina el Instituto de Enfermedades Infecciosas "Dr. Carlos Malbrán" ha informado en el período 2001-2003 el diagnóstico de 15 casos neonatales, 8 en niños y 20 en adultos 19,20.
6) Herpes simple perinatal El virus del herpes simple es ubicuo y está altamente adaptado al anfitrión, puede causar una amplia variedad de enfermedades. Existen 2 tipos el 1 (VHS-1) y el 2 (VHS-2). Son muy parecidos pero con epidemiología diferente, el VHS-1 es transmitido principalmente por saliva infectada, mientras que el HSV-2 es trasmitido sexualmente o a través del tracto genital infectado de la madre al recién nacido. La infección por VHS ha incrementado su prevalencia a lo largo del mundo en las últimas dos décadas. Se caracteriza por su neurovirulencia, su latencia y su capacidad de reactivación las cuales facilitan la permanencia de la endemia. La diseminación de la infección se produce en pacientes que tienen alterada la inmunidad de las células T (receptores de transplantes orgánicos, HIV+, etc.).
Es altamente prevalente, de esa manera la mayoría de los individuos muestra evidencias de infección por VHS. Respecto a la seroprevalencia se observa que los ACs contra HSV-1 aumentan con la edad comenzando en la niñez y en correlación con el nivel socioeconómico. A la edad de 30 años el 50% de los individuos de nivel socio-económico alto y el 80% de nivel más bajo son seropositivos. Los ACs contra el HSV-2 aparecen en la pubertad, relacionados con el grado de actividad sexual, más tarde en la vida la seroprevalencia puede ser del 20-80%. No hay evidencias claras que indiquen que el riesgo de la infección por VHS esté relacionado con la raza.
La infección por el VHS-1 transmitida por saliva es frecuente en los niños, aunque la primoinfección herpética puede ser observada a cualquier edad. Las infecciones por VHS 2 están agrupadas en el momento del parto (perinatal) y cuando comienza la actividad sexual, principalmente. La morbilidad y la mortalidad de la infección por VHS están relacionadas con complicaciones, entre las cuales citamos a continuación:
a) Infecciones bacterianas y fúngicas que no son frecuentes, como candidiasis balanoprepucial o vaginitis (el 10% de las mujeres afectadas de herpes genital primario, particularmente si además tienen diabetes, asocian candidiasis).
b) Infecciones oculares, no infrecuentes en niños, resultado de autoinoculación durante episodios de gingivoestomatitis herpética aguda o infección herpética asintomática orofaríngea. Principalmente son a HVS-1, salvo, en neonatos que son HVS-2. En el 25% de los casos es recidivante. Las úlceras repetidas es la causa principal de ceguera en USA.
c) Las infecciones en piel ofrecen las siguientes complicaciones:
I- eccema herpético (localizado o generalizado).
II- panadizo herpético.
III-herpes del gladiador: lesiones cutáneas por HSV-1 diseminadas han sido vistas en luchadores expuestos a la saliva infectada durante un combate.
d) infecciones viscerales resultado de la viremia, siendo común el compromiso multiorgánico. La hepatitis es muy destacada en la infección VSH fulminante, puede asociar leucopenia, trombocitopenia y coagulación intravascular diseminada.
e) Complicaciones del SNC:
I -Meningitis aséptica: es aguda pero generalmente se trata de una meningitis linfocítica benigna. En una serie estudiada, el 36% de las mujeres y el 13% de los hombres con HSV-2 genital primario tenía signos y síntomas meníngeos. HSV-1 demostrado con reacción en cadena de la polimerasa (PCR) en el LCR de pacientes con meningitis recidivante linfocítica benigna ha sugerido que el responsable es este virus más que un origen idiopático.
II -Ganglionitis y mielitis con posible retención urinaria, neuralgia o anestesia sacra.
III -El herpes simple origina en USA el 10- 20% de las causas de esta encefalitis.
IV-Durante el embarazo la mujer puede padecer infección por VHS ampliamente diseminada con una alta tasa de mortalidad neonatal (50%). La infección herpética en el tercer trimestre del embarazo está asociada con infección neonatal por VHS, retardo del crecimiento intrauterino y nacimiento de prematuros.
f) Herpes neonatal El 90% de las infecciones herpéticas son adquiridas en el momento del parto (perinatal), 5-8% son adquiridas in útero (congénitas), y unas pocas adquiridas después del parto (postnatal o neonatal). Esta últimas son causadas por contacto con secreciones genitales infectadas.
En el 70% de las madres la infección es asintomática. El riesgo de transmisión de una madre con infección primaria está cerca del 50%. Los neonatos y recién nacidos (de menos de 6 semanas de vida) tienen una muy alta incidencia de infecciones viscerales y del SNC. Sin tratamiento, la tasa de mortalidad es del 65%, existiendo un alto grado de secuelas neurológicas.
La enfermedad puede estar confinada a la piel, ojos o boca, o puede manifestarse como encefalitis o enfermedad diseminada visceral, comprometiendo pulmones, hígado, corazón, suprarrenales y la piel 21.
7) Varicela neonatal El resultado de la infección en útero del virus varicela/zóster varía según el momento del embarazo, de la siguiente manera:
I- Síndrome de la varicela congénita Este ocurre en el 2% de los niños nacidos de mujeres con varicela durante el primer o segundo trimestre del embarazo. Estos neonatos presentan retardo del crecimiento, microcefalia, atrofia cortical, hipoplasia de los miembros, microftalmia, cataratas, coriorretinitis, y cicatrices cutáneas.
II- Herpes zóster infantil Usualmente se manifiesta dentro del primer año de vida, su causa es la varicela materna contraída después de la semana 20 de gestación. Suele involucrar las dermatomas torácicas.
III- Varicela neonatal Puede ser una enfermedad seria, dependiendo de la relación temporal entre el momento del desarrollo de la varicela materna y el parto:
a) varicela materna desarrollada entre los 5 días antes y hasta 2 días después del parto, el feto está expuesto a la viremia secundaria de la madre, adquiere así el virus transplacentario pero no los ACs protectores pues aún la madre no los ha formado. En estas circunstancias la varicela probablemente sea severa y diseminada. Se requiere profilaxis o tratamiento con globulina inmune varicela-zóster (VZIG) y aciclovir. Sin estas drogas la tasa de mortalidad puede ascender al 30%. Las causas principales de muerte son neumonía y hepatitis fulminante.
b) varicela materna desarrollada antes de los 5 días previos al parto, la madre ha podido elaborar ACs que serán pasados al feto junto con el virus. Los neonatos a término de estas madres usualmente tendrán varicela moderada. La VZIG no es recomendada especialmente, pero el aciclovir puede ser usado. Cada lesión individual comienza como una mácula eritematosa que luego evoluciona por los estados de pápula, vesícula, pústula y costra. El enrojecimiento y/o el edema perilesional deben hacer sospechar sobreinfección bacteriana. En un niño con varicela, sin otra patología sobreagregada, usualmente la cantidad de elementos varía de 250-500 pero hay casos que van desde 10 elementos hasta 1500 22,23.
CONCLUSIONES
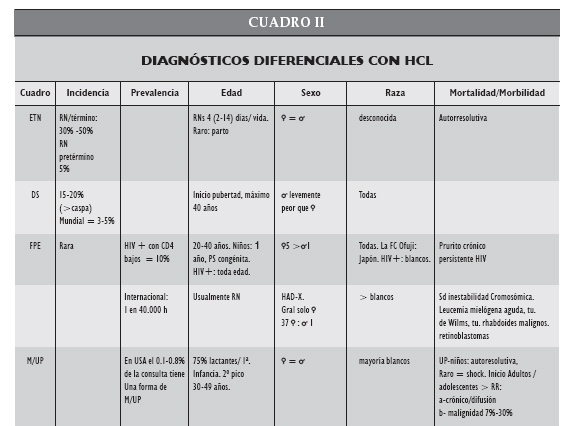
Debido al gran número de patologías sujetas a comparación, el escaso o nulo acceso a datos previos y la rareza de las mismas, es difícil hacer comparación entre ellas con estricto valor epidemiológico. Sin embargo, con los pocos datos disponibles, y tomándonos grandes licencias científicas, entre ellas cotejar datos de distintos países, hemos elaborado el Cuadro II, donde estas entidades han sido ordenadas de mayor a menor frecuencia.

1. Selden S. Seborrheic Dermatitis. 2005; http://www.emedicine.com/specialties.htm [ Links ]
2. Chang CH. Incontinentia Pigmenti. 2005; http://www.emedicine.com/specialties.htm [ Links ]
3. Hogan D y Lewis VP. Mastocytosis. 2002; http://www.emedicine.com/specialties.htm [ Links ]
4. De Andrea I, Castillo M y Walter T. Desarrollo psicomotor y conducta en lactantes anémicos por deficiencia de hierro. En: O'Donnell AM, Viteri FE Carnuega ES. Deficiencia de hierro: desnutrición oculta en América Latina. Publicación CESNI, Editorial Gaudian. Buenos Aires. Argentina. 1997; 18: 14-19. [ Links ]
5. Underwood B. Micronutrient Malnutrition: Is it being eliminated? Abstracts of The 16th International Congress of Nutrition. Montreal. Canadá. Abs. 16; 1997. [ Links ]
6. Delgado H y Mejía L. Taller transición nutricional en América Latina. Actas del Congreso Latinoamericano de Nutrición. Caracas. Venezuela. 2001; 16-19. [ Links ]
7. Selim A y Shea CR. Langerhans Cell Histiocytosis. 2005; http://www.emedicine.com /specialties.htm [ Links ]
8. Sullivan KE y col. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr 1994; 125: 876. [ Links ]
9. Dibbern DA y Routes JN. Wiskott - Aldrich syndrome. 2005; http://www.emedicine.com/specialties.htm [ Links ]
10. van Zander JE. Cutaneous Rosai-Dorfman disease. Dermatology Online Journal 2004; 10 (3): 12. [ Links ]
11. Schee MM y col. Sinus histiocytosis with massive lymphadenopathy. J Am Acad Dermatol 1997; 37: 643. [ Links ]
12. Horenstein MG y McNeil JP. Xanthomas. 2005; http://www.emedicine.com/specialties.htm [ Links ]
13. Shani-Adir A y Mancini AJ. Dermatology Quiz neonatal pustular melanosis. Children's Memorial Hospital. 2006. [ Links ]
14. Durham BA y Laumann A. Transient Neonatal Pustular Melanosis. 2005; http:/ /www.emedicine.com/specialties.htm [ Links ]
15. Ramamurthy RS, Reveri M, Esterly NB y col. Transient neonatal pustular melanosis. J Pediatr 1976; 88 (5): 831-835. [ Links ]
16. Ta A y Sandler B. Common transient neonatal dermatoses. En : Harper J, Oranje A y Prose N. Textbook of Pediatric Dermatology. United Kingdom. Blackwell Science. 2000; 59-61. [ Links ]
17. Scheinfeld NS y Lambiase MC. Candidiasis cutánea. 2006; http://www.emedicine.com [ Links ]
18. Larralde de Luna M. Dermatosis neonatales. En Dermatología neonatal y pediátrica. Editorial EDIMED. Buenos Aires. Argentina. 1995: 22-29. [ Links ]
19. Cisternas VA, Lagos NN, Galstuch LJ, González RC, Claudio García CC y Díaz TJ. Infección por Listeria monocytogenes y embarazo con buen resultado perinatal. Rev Chil Obstet Ginecol 2002; 67 (3): 237-241. [ Links ]
20. Lutwick LI y Seenivasan M. Herpes simple. 2005; http://www.emedicine.com [ Links ]
21. Metha PN y Chatterjee A. Varicella. 2005, http://www.emedicine.com. [ Links ]
22. Centers for Disease Control and Prevention (CDC): National, state, and urban area vaccination coverage among children aged 19-35 months-United States, 2004. MMWR Morb Mortal Wkly Rep 2005; 54 (29): 717-721. [ Links ]
23. Zhou F, Harpaz R, Jumaan AO y col. Impact of varicella vaccination on health care utilization. JAMA 2005; 294 (7): 797- 802. [ Links ]

