Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El termino ictiosis define como un grupo de enfermedades con alteraciones en la cornificación, su nombre deriva de la palabra griega icthysque significa pez, debido al aspecto escamoso de la piel, 1 esta patología se define como un grupo de enfermedades que producen escamas visibles en toda o gran parte de la superficie de la piel, se caracterizan por presentar hiperqueratosis y/o descamación.1 Estas anormalidades ocurren en cualquiera de las fases que se llevan a cabo durante la diferenciación epidérmica terminal, cuya finalidad es la de formar el estrato córneo.2
La ictiosis hereditaria corresponde a trastornos genéticos de la cornificación, a diferencia de la ictiosis adquirida, que pueden ser secundarias a neoplasias malignas, enfermedades autoinmunes, inflamatorias, infecciosas, metabólicas, reacciones medicamentosas y deficiencias nutricionales.1 En el año 1933, Cockayne sugiere la primera clasificación genética, donde se distinguen formas autosómicas (dominante- recesiva) y la forma ligada al sexo, más tarde en 1965, Wells y Kerr introducen las formas de herencia entre la ictiosis vulgar y la ictiosis ligada al X, para el año 1966, Frost y colaboradores distinguen 2 grupos de ictiosis: normocinéticas e hipercinéticas, las cuales dependerán si el defecto se encuentra a nivel del proceso de cornificación o en el tiempo durante el cual se lleva a cabo la descamación, por último en 1978, Kopp y colaboradores evidencian el déficit de la actividad de la sulfatasa esteroidea.3
En el año 2009 un grupo de expertos desarrolló una nueva clasificación de consenso basada principalmente en las características clínicas, considerando los aspectos fisiopatológicos y moleculares descubiertos hasta el momento, lo que ha permitido facilitar la comprensión de la enfermedad y el estudio de los pacientes. Esta clasificación identifica 36 tipos de ictiosis, las cuales se dividen en subgrupos de acuerdo a la presencia o no de compromiso extracutáneo, frecuencia de la enfermedad y patrón de herencia.1 (tabla 1). Cabe destacar que las manifestaciones extracutáneas permiten identificar 2 grandes grupos de ictiosis, cuando el defecto genético se manifiesta exclusivamente en la piel se denominan ictiosis no sindrómicas, mientras que la presencia de alteraciones en otros órganos o sistemas además de la piel, define las ictiosis sindrómicas.1,4 (tabla 2-3)
La ictiosis lamelar tiene una incidencia de 1 en 200,000 o 300,000 nacimientos,5afecta ambos sexos y a todas las razas, sin un patrón geográfico definido; sin embargo, debido a que generalmente presenta un patrón autosómico recesivo se observa más en países con alto índice de consanguinidad; aunque existen algunas comunicaciones de casos transmitidos con un patrón de herencia autosómico dominante.2 Existen pocos datos sobre la epidemiología de las ICAR. En Estados Unidos se ha estimado una prevalencia al nacimiento de 1:100.000 para la IL y 1:200.000 para la EIC. Otros estudios han calculado una prevalencia conjunta para IL y EIC de 1:200.000-300.000. En algunos países, como Noruega, la prevalencia estimada es mayor (1:91.000) debido a la existencia de mutaciones fundadoras.5
Este padecimiento se desarrolla debido a una mutación del gen de la transglutaminasa 1, que es una proteína asociada a la membrana de los queratinocitos, de 92 kDa; el subtipo mayor se encuentra en la epidermis y es la responsable de ensamblar las proteínas precursoras que forman la capa córnea, la banda marginal se produce por la síntesis de loricrina e involucrina; formadas por la participación de la transglutaminasa de los queratinocitos (TG). El gen de TG se localiza en el cromosoma 14q11 y otros en el cromosoma 17p13.1, identificados como ALOXE3 y ALOXE12B; recientemente se han comunicado casos con mutaciones en el cromosoma 2.2 sin embargo en casos de Ictiosis severa la herencia es autosómica recesiva, y al igual que la anterior la mayoría de los casos son producto de mutaciones en el gen que codifica la enzima transglutaminasa-1 (TGM-1), con mutaciones en otros genes como NIPAL4, CYP4F22, ABCA12 y otros aún no identificados.1,5 La disminución o pérdida de función de la enzima TGM-1 altera la formación de la envoltura cornificada y la unión de los lípidos intercelulares, resultando en un trastorno importante de la diferenciación y descamación, generando hiperqueratosis por retención.1,5
En 1985 Sibert y colaboradores definieron la ictiosis como un defecto en la síntesis de una proteína del estrato córneo, filagrina, y de su precursor, profilagrina, en pacientes con esta patología. La filagrina interactúa con la queratina para formar los filamentos de filagrina-queratina en el estrato córneo, también se produce la liberación de aminoácidos libres, manteniendo la hidratación de los corneocitos y una descamación adecuada, en la ictiosis se altera la profilagrina, causando disminución de la filagrina, aminoácidos libres, lo que trae como consecuencia la descamación (por retención figura 1).3, 6

La ictiosis congénita recesiva tiene una prevalencia estimada de 1:138.000-1:300.000.1,4 y el 60% de los casos corresponde a la manifestación inicial de eritrodermiaictiosiforme congénita o ictiosis lamelar,1,7se dividieron en 5 subtipos en 1992, se desarrolla la clasificación de Heidelberg:6
Ictiosis congénita tipo I: Evoluciona a EICnoB; inicia como bebé colodión que posteriormente progresa a eritrodermia con escamas en tronco, cara y partes flexoras de las extremidades.6
Ictiosis congénita tipo II: Evoluciona a ictiosis laminar. Se presenta como un bebé colodión al nacer, con importante eclabión y ectropión, confundida frecuentemente con el feto arlequín. La microscopía electrónica demuestra hendiduras de colesterol en el estrato córneo.6
Ictiosis congénita tipo III: No desarrolla eritrodermia, se observan escamas con patrón reticulado. La microscopía electrónica demuestra membranas perinucleareselongadas en las capas granulosas y córneas.6
Ictiosis congénita tipo IV: Se presenta en bebés prematuros y se observan malformación de orejas, piel con aspecto de «parchado» e hiperqueratosis palmo-plantar. Es común la muerte prenatal debido a la obstrucción de la tráquea que conduce a asfixia. La microscopía electrónica revela acúmulos de membranas de lípidos y edema paranuclear en las capas granulosa y córnea.6
Ictiosis congénita tipo V: Inicia como bebé colodión. Sólo se ha reportado un caso de este tipo de ictiosis, con escama generalizada en cara, tronco y extremidades, eritrodermia pronunciada, hiperqueratosis palmoplantar, uñas disqueratósicas, hipospadias, hipogonadismo y retardo en el crecimiento. La microscopía electrónica muestra marcada supresión de la queratinización y células binucleadas.6
Las características clínicas se manifiesta al nacer como bebé colodión, esta membrana que los recubre se desprende dentro de los 10 a 14 días después de nacidos.2,8 Posteriormente se observan escamas grandes romboidales, hiperpigmentadas, oscuras, gruesas y con fisuras, de distribución generalizada y patrón en mosaico, con mínima eritrodermia o sin ella, la hiperqueratosis generalizada y el taponamiento de los conductos sudoríparos, inducen la disminución de la sudoración e hipertermia, así mismo existen alteraciones en las glándulas sudoríparas que favorecen una piel seca y no flexible, con disminución de los arcos de movilidad articular con contracturas flexurales; la queratodermiapalmoplantar es frecuente, las uñas pueden presentar onicogrifosis, desviación lateral, hiperqueratosis subungueal, surcos y fisuras longitudinales, las alteraciones en el tallo del pelo no son frecuentes, suele presentar ectropión, eclabión, hipoplasia de cartílagos nasales y auriculares.2,6
Histológicamente se caracteriza por moderada hiperqueratosis ortoqueratósica masiva, disminución o ausencia del estrato granuloso, observado hasta en el 75% de los pacientes o hipergranulosis y acantosis moderadas;2 disminución de los anexos, hiperqueratosis folicular con tapones córneos foliculares, en dermis existe vasodilatación e infiltrado moderado linfohistiocíticoperivascular, en algunos casos pueden presentarse con patrón psoriasiforme.5 La tasa de proliferación celular es normal; El diagnóstico diferencial se realiza con ictiosis ligada a X, eritrodermiaictiosiforme congénita y síndrome de Netherton.1
El diagnóstico, en el grupo más frecuente no genera dificultad, si realizamos una buena historia, determinando el árbol genealógico y los datos durante el examen físico completo, si existen anomalías asociadas, que pueden orientar hacia determinados tipos de ictiosis infrecuentes.3 Es importante realizar una historia clínica detallada y examen físico completo con énfasis en el fenotipo cutáneo (patrón de descamación e hiperqueratosis, calidad y color de las escamas), inicio y evolución (bebe colodión, eritrodermia al nacimiento o ictiosis de inicio tardío), presencia de otras características dermatológicas (eritema, prurito, ampollas, erosiones, alteración de faneras), compromiso extracutáneo e historia familiar (patrón de herencia).1,2 Los hallazgos clínicos e histológicos aportan información relevante para el diagnóstico.1 En el caso de ictiosis vulgar, la reducción o ausencia del estrato granuloso es característico y en la ictiosis epidermolítica, la presencia de ampollas microscópicas y fisuras acantolíticas orientan al diagnóstico, aun cuando no se observen erosiones o ampollas. La observación de pelos a la microscopia también aporta datos interesantes. La alteración en tallo de bambú o tricorrexisinvaginata es un criterio diagnóstico de síndrome de Netherton.3En cambio, la visualización de pelos atigrados con luz polarizada es característica de tricotiodistrofia, la disminución de gránulos de queratohialina es común en la ictiosis vulgar, y la disrupción del citoesqueleto, separación de desmosomas, secreción anormal de cuerpos lamelares se observa en las ictiosis queratinopáticas.1, 3
El tratamiento sintomático debe ser individualizado, ya que la efectividad y tolerancia de cada paciente es diferente, es importante considerar la edad, el tipo y gravedad de la ictiosis, la extensión y/o localización de las lesiones y la respuesta a terapias previas. El dermatólogo tiene un rol fundamental en el diagnóstico, tratamiento y seguimiento, sin embargo, el manejo debe ser realizado por un equipo multidisciplinario.5,8
Los emolientes y queratolíticos tópicos suelen ser la primera línea de tratamiento, ya que mejoran la función de barrera y facilitan la descamación al ser aplicados al menos 2 veces al día, para hidratar la piel se utilizan preparados con urea, glicerol o vaselina.1, 9 En pacientes con escamas gruesas e hiperqueratosis marcada se puede añadir uno o más agentes queratolíticos como urea en altas concentraciones, ácido láctico, ácido salicílico y propilenglicol.1,5 Los moduladores de la diferenciación de queratinocitos, como retinoides tópicos (tretinoína, adapaleno y tazaroteno) pero pueden causar irritación local.1,5,9 Sin embargo a pesar que los retinoides orales son particularmente eficaces en la reducción de la cantidad de escama, pueden exponer al paciente a una eritrodermia subyacente.2
Los retinoides orales (isotretinoína y acitretin) son derivados de la vitamina A que actúan regulando la proliferación y diferenciación celular, previniendo la hiperqueratosis y facilitando la descamación. En general, se reservan para ictiosis graves o refractarias al tratamiento tópico. Se recomienda iniciar con dosis bajas, y mantener un seguimiento estrecho por sus importantes efectos adversos (alteraciones mucocutáneas, teratogenicidad, alteraciones musculoesqueléticas, metabolismo lipídico y hepático).5, 1
Un estudio preliminar desarrolló un protocolo de investigación con la utilización de Isotretinoina, tomando como base el PASI (Psoriasis Area and SeverityIndex) que cuantifica la extensión y la intensidad en el marco de la severidad de la Psoriasis, el cual modificamos tomando los mismos criterios y le denominamos IASIS (ICHTHYOSIS AREA AND SEVERETY INDEX), tomando parámetros como la extensión, eritema, descamación, y adhesión de las escamas, además de evaluar el comportamiento del ectropión (eversión del párpado), dicho protocolo se basa: en el uso de urea 10%, lagrimas artificiales y shampoo con piritionato de Zinc 3 veces a la semana para el cuero cabelludo asociado a aceite de oliva. Los controles se realizaron mensualmente y los exámenes de laboratorios cada 2 meses, la dosis fue calculada de acuerdo al peso y edad, <5 años 5 mg diarios VO después de almuerzo, se absorbe mejor con los alimentos; 6-12 años 10 mg y > de 15 años 20 mg cada día, los resultados mostraron la disminución de acuerdo a las semanas de tratamientos, mejorando su IASI, obteniendo una respuesta excelente mejorando la adherencia de las escamas, pliegues palmo-plantares, ectropión en un 50 % sin alteraciones de importancia, salvo la xerosis cutánea, con aumento importante de la descamación, a pesar de los resultados los retinoides orales deben reservarse para casos con ictiosis graves.4 Los derivados de vitamina D (calcipotriol), son especialmente útiles en áreas hiperqueratósicas.1,5
Los preparados con N-acetilcisteína al 10% en una base water in oil(w/o), han demostrado ser efectivos, seguros y bien tolerados en pacientes con IL,1, 10 en los neonatos y lactantes, se recomienda utilizar un vehículo sin medicación, dado que la piel es más sensible, menos tolerable a los queratolíticos y presenta un mayor riesgo de absorción percutánea de productos tópicos como urea y ácido salicílico, entre otros. Respecto a la selección de tratamiento, es importante considerar que las ICAR responden bien al uso de queratolíticos potentes, mientras que las ictiosis queratinopáticas tienden a empeorar con su uso, en caso de ectropión, el uso de lágrimas artificiales, lubricantes oculares y la hidratación del rostro disminuyen la xeroftalmia y la retracción palpebral, por lo que la cirugía se reserva para casos de difícil manejo.1
FUTURAS TERAPIAS
La terapia génica ha surgido como una posible alterativa curativa en el tratamiento de las ictiosis, basándose en la corrección de los genes defectuosos. Estudios experimentales en ictiosis severas, como IA e IL, muestran resultados prometedores. En IA se ha logrado corregir las mutaciones en el gen ABCA12, recuperando la expresión fenotípica de queratinocitos en cultivos celulares, mientras que en IL se ha conseguido restaurar la enzima TGM-1, corrigiendo la expresión fenotípica de piel humana trasplantada en ratones inmunosuprimidos.1 Cabe esperar que los avances en el conocimiento de su etiopatogenia lleven a permitir solucionar el defecto genético o sustituir las proteínas deficitarias, pero son muy pocos los ensayos controlados que hacen referencia al tratamiento de las ictiosis y así el registro Cochrane sólo recoge 13 referencias bibliográficas, todas ellas sobre tratamientos tópicos.2
CASO CLINICO
Paciente pre-escolar de sexo femenino, de 4 años de edad, natural y procedente del estado Portuguesa, Venezuela. Fototipo cutáneo 3/6 según la escala de Fitzpatrick, quien acude al servicio de Dermatología de la Ciudad Hospitalaria Dr. Enrique Tejera CHET Valencia, estado Carabobo, por presentar piel seca con escama gruesa oscura desde los 2 meses de vida. Producto de madre de 33 años de edad primigesta, embarazo controlado, serología HIV, VDRL, IgM toxoplasmosis negativas en los tres trimestres, uroanálisis y urocultivo no patológicos, complicado con HTA Gestacional controlado con metildopa vía oral orden diaria, obtenido por parto eutócico simple a las 38 semanas por crisis hipertensiva, se obtiene recién nacido a término TAN: 52 cm PAN: 3,200 kg, complicado con síndrome de dificultad respiratoria, hipoxia neonatal, es hospitalizada durante 48 horas en neonatología bajo diagnóstico de asfixia perinatal severa, síndrome metabólico: hipoglicemia, por lo que ameritó reanimación cardiopulmonar, posteriormente O2 por oxihood 12 horas y posteriormente por manguera corrugada e hidratación parenteral, con evolución satisfactoria es egresada sin complicaciones. Desarrollo psicomotor acorde a edad sin alteraciones, antecedentes familiares niegan consanguinidad y antecedentes similares en la familia. Durante el periodo neonatal presenta eritrodermia y a los dos meses de edad presenta dermatosis generalizada a predominio facial y de extremidades caracterizada por descamación, acude a facultativo privado quien indica tratamiento tópico sin mejoría clínica, empeorando cuadro clínico.Acude a nuestro servicio a los 3 meses de edad por presentar dermatosis generalizada, bilateral y simétrica caracterizadapor escama de tamaños variables poligonales, gruesa, adherente, hiperpigmentada, acompañada de xerosis y algunas fisuras (figuras 5-6-7). Sin alteraciones de palmas y plantas (figura 3). En el resto de la piel y los anexos se aprecia ectropión (figuras 1-2) escamas en piel cabelluda, pelo seco y quebradizo, alteraciones unguealesonicodistrofia, pitting y onicorrexis (figura 4), hipohidrosis con intolerancia al calor. A la dermatoscopia se evidencia escamas poligonales, hiperpigmentadas, gruesas adherentes con patrón en mosaico, además se observan fisuras de tamaños variables. (figura 8)
Se realizó el diagnóstico clínico de Ictiosis Laminar Congénita Autosómica Recesiva,ademásde valoración por el servicio de genética con realización de pedigreegenético evidenciando en la Genealogía un caso segregante de Ictiosis LamelarAutosómica Recesiva con caso índice en la III generación y un riesgo de recurrencia del 25% en cada embarazo no se realiza test genético en vista de no estar disponible en Venezuela.
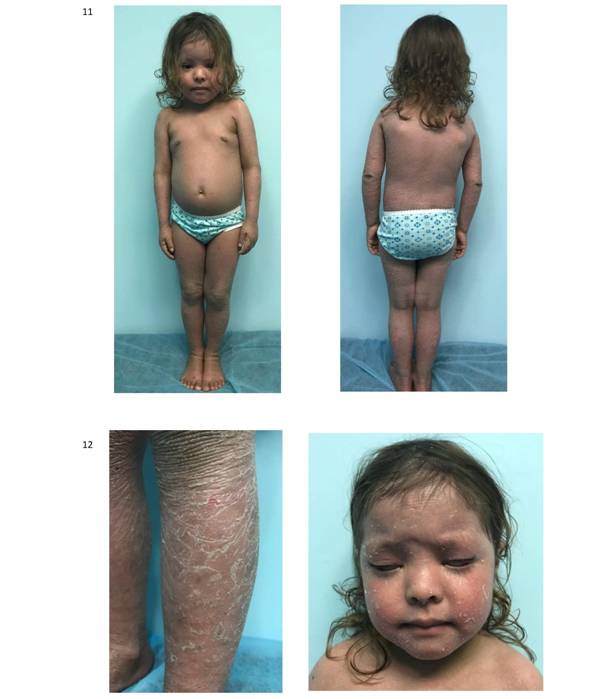
Como parte de la valoración integral y tratamiento se da explicación amplia de la enfermedad; manejo sintomático a base de emolientes y shampoo alquitranado, syndet, aceite de oliva ozonificado, SOFT esteroides, vitamina D, ácido fólico, Zinc, referida al servicio de pediatría y puericultura quien indica dieta balanceada (hipoalergénica - hiperproteica- normocalórica), con evolución satisfactoria (figura 11-12) evolución con terapia combinada (figura 13) valoración por el departamento de genética quienes con los datos obtenidos elaboranpedigree (figura 10). Actualmente se encuentra en rehabilitación; se solicitó interconsulta a oftalmología en donde indican tratamiento con ungüento y gotas lubricantes.
Laboratorios: hematología completa leucocitos: 8.200 mm3, Neutrófilo: 60%, Eosinófilos: 0%, Hemoglobina: 12gr/dl, Glicemia 80mg/dl, Urea 21 mg/dl, Creatinina sérica0,7 mg/dl TGO 18 U/L TGP 20 U/L ecogramaabdomino-pélvico sin alteraciones.
Hallazgos histopatológicos: hiperqueratosis con formación de vesículas intercorneales, espongiosis y acantosis moderada, tapones córneos foliculares e infiltrado moderado perivascular (figura 9).
COMENTARIO
La ictiosis autosómica recesiva es un término que abarca la Ictiosis Laminar (IL) esta se subdivide en 5 subtipos6 explicados anteriormente, en vista de la evolución de nuestro caso podemos determinar que se trata de una patología autosómica recesiva subtipo II que evolucionó a Ictiosis Laminar, generalmente se presenta como un bebé colodión al nacer, pero la clínica que presentó nuestra paciente inició como una eritrodermia con mejoría clínica espontánea y reapareció a los 2 meses de edad presentando descamación y evolucionando posteriormente a escamas, oscuras, grandes, romboidales, con algunas fisuras, asociándose además a alteraciones en las glándulas sudoríparas que favorecen una piel seca disminuyendo su flexibilidad. En el caso que presentamos, desafortunadamente, a pesar de haberse iniciado de esta forma, y desarrollar manifestaciones clínicas floridas de la enfermedad, no se pudo determinar cuál era la mutación genética especifica ya que actualmente no se cuenta con los medios para realizar dicho estudio en nuestro país, sin embargo,las terapias tópicas combinadas y el manejo multidisciplinario nos permitieron obtener una mejoría clínica satisfactoria, a pesar de las complicaciones propias que ha desarrollado la paciente durante la evolución natural de la enfermedad, con el desarrollo de ectropión; si bien es cierto, esta es una enfermedad no curable, pero se puede realizar una intervención médica adecuada, principalmente con el objeto de disminuir al mínimo las secuelas.
DISCUSIÓN
La nueva clasificación de ictiosis nos permite una mejor comprensión de la enfermedad y un correcto diagnóstico, ya que considera los aspectos clínicos, histológicos y moleculares de esta enfermedad. Dentro de la variedad de presentaciones clínicas de ictiosis lamelar autosómica recesiva, no es frecuente y de difícil tratamiento. En el Servicio de Dermatología de la ciudad hospitalaria Dr. Enrique Tejera CHET en Carabobo, Venezuela, la presentación clínica de esta patología es baja, correspondiendo el 60% de los casos a manifestación inicial de eritrodermiaictiosiforme congénita o ictiosis lamelar,1,7tendencia relacionada con resultados de otros estudios internacionales. Se ilustra el caso de pre-escolar femenina con clínica e histopatología demostrativa de una variante de ictiosis congénita recesiva, quien recibió tratamiento combinando tópico, sistémico, y análogos de la vitamina D, evolucionando satisfactoriamente a las semanas de tratamiento.
El uso de emolientes y queratolíticosfavorecen la eliminación de la escama y el uso de sustancias como aceites vegetales,2 como oliva, ozonificados, a nivel cutáneo promueve la proliferación de queratinocitos y fibroblastos, modula la expresión del factor nuclear κβ, y otros marcadores pro inflamatorios, factores de crecimiento. Lo anterior se traduce en un efecto antiinflamatorio, una regeneración de la matriz intercelular y un incremento en la síntesis de colágeno y elastina. Además induceuna actividad remodeladora y antimicrobiana, y potencializa el efecto terapéutico de múltiples principios activos utilizados por vía tópica.12
El Ozono, es una molécula obtenida a partir de oxígeno medicinal sometido a un gradiente eléctrico, la ozonización de los aceites vegetales se basa en el burbujeo; durante esta reacción se forman diferentes sustancias como: lipoperóxidos, ozónidos, aldehídos, cetonas. Estos ácidos grasos presentes en los aceites vegetales desde el punto de vista químico son ácidos carboxílicos con una cadena alifática que puede ser monoinsaturada o tener varias insaturaciones, en el caso del aceite de oliva tiene predilección a ácidos grasos mono insaturados.12
El mecanismo de acción exacto sobre la actividad biológica de los aceites ozonizados es aún desconocido. Sinembargo existen varias hipótesis. Una de ellas plantea que es probable que los triozonidos estables al entrar en contacto con los exudados de las heridas que se encuentran a una temperatura de aproximadamente 37ºC, se descompongan y generen ozono, formando peróxido de hidrógeno y lipoperóxidos, que serían los responsables de los efectos regenerativos y desinfectantes. Se plantea que la liberación lenta de ozono en las heridas favorece el proceso de cicatrización, no solo por la desinfección local sino también por favorecer a nivel local la liberación de citocinas con efectos reparadores; 12,13con relación a los efectos antimicrobianos el aceite ozonizado en contacto con un microorganismo genera afectaciones en su citoplasma; además de originar una reducción en el contenido de ácidos nucleicos que se corresponde con una reducción de la actividad lipasa, amilasa, keratinasa y ureasa.12
La liberación de factores de crecimiento como PDGF, TGF-β y VEGF pueden incidir en la remodelación tisular, sin embargo, el efecto de oxidación local en los tejidos por los componentes del aceite ozonizado pueden estimular mecanismos antioxidantes endógenos y promover la reparación de los tejidos, esto se ha demostrado en múltiples estudios donde se evaluó la eficacia de la aceleración del proceso de cicatrización inducida por el aceite ozonizado, donde se involucran los factores de crecimiento; por lo que la activación de los mecanismos de reparación de los tejidos es de gran utilidad para el tratamiento tópico en varias patologías de índole dermatológico.12
En base a estos resultados y los reportados en otros casos clínicos de ictiosis lamelar, 11se indica el uso tópico de aceite de oliva ozonificado en este caso de ictiosis lamelar obteniendo una reparación del tejido y evidenciando una evolución sastisfactoria, asimismo es de vital importancia usar terapias combinadas que nos permitan potenciar la eficacia de nuestro tratamiento como los derivados de la vitamina D, cuyas funciones autocrinas inhiben la progresión celular y promueven diferenciación y apoptosis a través de receptores en múltiples estirpes celulares (eficacia respaldada en múltiples estudios).Estas nuevas terapias representan una herramienta terapéutica del presente y el futuro en diferentes estados patológicos de la piel.

El siguiente cuadro nos refleja las características de los diferentes tipos de ictiosis con énfasis en los tipos más clásicos.





AD: autosómica dominante, AR: autosómica recesiva; ARC: artrogriposis-disfunción renal-colestasis; IFAP: ictiosis folicular-atriquiafotofobia; IH: ictiosis-hipotricosis; IHCE: ictiosis-hipotricosis-colangitis esclerosante; RN: recién nacido; XD: dominante ligada al cromosoma X; XR: recesiva ligada al cromosoma X.


Figura 4 Alteraciones unguealesonicodistrofiapitting, onicorrexis, además se evidencia escamas poligonales finas, adherentes y nevo de unión de aproximadamente 0,5 cm de diámetro sin patrón a la dermatoscopia.


Figuras 6 Dermatosis bilateral y simétrica caracterizada por escama de tamaños variables poligonales, gruesa, adherente, hiperpigmentada, acompañada de xerosis y algunas fisuras.

Figuras 7.Dermatosis bilateral y simétrica caracterizada por escama de tamaños variables poligonales, gruesa, adherente, hiperpigmentada, acompañada de xerosis y algunas fisuras.

Figura 8 Dermatoscopia de contacto y electrónica, donde se evidencian escamas poligonales, hiperpigmentadas, gruesas adherentes con patrón en mosaico, además se observa fisuras de tamaños variables.

Figura 9.Corte histopatologico teñido con H-E a un aumento de 10-40x hiperqueratosis laminar compacta, acantosis y hipergranulosis.