










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInsuficiencia cardíaca
versión On-line ISSN 1852-3862
Insuf. card. vol.5 no.4 Ciudad Autónoma de Buenos Aires oct./dic. 2010
ARTÍCULO DE REVISIÓN
Hipertiroidismo y sistema cardiovascular
Bases fisiopatológicas y su manifestación clínica
Diego Mantilla1, Mónica Liliana Echin2, Cecilia Perel3
1 Médico residente de cardiología. Hospital "Eva Perón" (Ex Castex). San Martín. Buenos Aires. República Argentina.
2 Médica endocrinóloga. Especialista en endocrinología ginecológica. Jefa del Servicio de Endocrinología del Hospital "Bernardino Rivadavia". Ciudad de Buenos Aires. República Argentina.
3 Médica cardióloga. Especialista en hipertensión arterial. División cardiología. Hospital "Bernardino Rivadavia". Ciudad de Buenos Aires. República Argentina.
Correspondencia: Dr. Diego Mantilla
Mateu 3354 Piso 1º "B". CP: 1650. San Martin. Buenos Aires. República Argentina.
Tel.: (54-11) 4752-2715.
E-mail: diegomanti2003@yahoo.com.ar
Recibido: 28/04/2010
Aceptado: 23/09/2010
Resumen
La glándula tiroides y el corazón están estrechamente relacionados fisiológicamente, concepto reforzado por los predecibles cambios de la función cardiovascular en los distintos trastornos tiroideos. Para entender, diagnosticar y tratar las cardiopatías que acompañan al hipertiroidismo, es importante conocer bien los mecanismos celulares de la acción de las hormonas tiroideas (HT) sobre el corazón y sobre las células del músculo liso.
El objetivo de esta revisión es profundizar en los mecanismos de acción de la HT sobre el corazón y sistema vascular periférico, los cambios anátomo-estructurales que se producen a nivel miocárdico, la interacción con el sistema nervioso simpático, los trastornos en la actividad eléctrica, las alteraciones hemodinámicas sobre la contractilidad miocárdica y la relajación ventricular. Las manifestaciones clínicas del hipertiroidismo pueden presentar síntomas iniciales como palpitaciones hasta avanzar a un estado de mayor complejidad como es la insuficiencia cardíaca (IC), entidad de alta morbimortalidad. Esta última puede manifestarse con alteraciones del ritmo, deterioro en la función ventricular y afectación valvular, que dejadas a su evolución natural pueden llevar a la muerte del paciente, dando relevancia al reconocimiento temprano por parte del profesional de la probable etiología hormonal de la IC y/o como causa de su descompensación. Este diagnóstico permite realizar un tratamiento precoz y eficaz, ya que estados de eutiroidismo llevan a la normalización de los trastornos cardiovasculares en la mayoría de los pacientes.
Palabras clave: Hipertiroidismo; Hormona tiroidea; Sistema cardiovascular; Fibrilación auricular; Insuficiencia cardíaca; Daño miocárdico; Hipertensión pulmonar
Summary
Cardiovascular system and hyperthyroidism
Pathophysiological bases and its clinical manifestation
The thyroid gland and the heart are closely related physiologically, this concept is reinforced by the predictable change in cardiovascular function in various thyroid disorders. To understand, diagnose and treat heart diseases that accompany hyperthyroidism is important to understand the cellular mechanisms of action of thyroid hormones (TH) on the heart and smooth muscle cells.
The aim of this review is to reinforce the mechanisms of action of TH on the heart and peripheral vascular system, anatomic and structural changes that occur at myocardial level, interaction with the sympathetic nervous system, impaired electrical activity, hemodynamic changes on myocardial contractility and ventricular relaxation. The clinical manifestations of hyperthyroidism can lead to symptoms such as palpitations initial advance to a state of greater complexity such as heart failure (HF), an entity of high morbidity and mortality, could be expressed with rhythm disturbances, damage and impaired ventricular function valve, which left to their natural evolution may lead to death of the patient, giving relevance to the early recognition by professionals of the hormonal etiology of HF and/or the cause of his decompensation. This diagnosis allows early treatment and effective as euthyroid states lead to the normalization of cardiovascular disease in most patients.
Keywords: Hyperthyroidism; Thyroid hormone; Cardiovascular system; Atrial fibrillation; Heart failure; Myocardial damage; Pulmonary hypertension
Resumo
Hipertireoidismo e sistema cardiovascular
Bases fisiopatológicas e sua manifestação clínica
A glândula tireóide e coração estão intimamente relacionados fisiologicamente, um conceito reforçado pela previsíveis alterações da função cardiovascular em várias doenças da tireóide. Para entender, diagnosticar e tratar das doenças do coração, que acompanham o hipertireoidismo, é importante compreender os mecanismos celulares de ação dos hormônios tireóideos (HT) no coração e células musculares lisas.
O objetivo desta revisão é o de reforçar os mecanismos de ação dos HT sobre o coração e sistema vascular periférica, anatômico e variações estruturais que ocorrem a nível do miocárdio, a interação com o sistema nervoso simpático, alteração da atividade elétrica alterações hemodinâmicas na contratilidade miocárdica e relaxamento do miocárdio ventricular. As manifestações clínicas do hipertireoidismo podem levar a sintomas como palpitações adiantamento inicial para um estado de maior complexidade, tais como insuficiência cardíaca (IC), uma entidade de alta morbidade e mortalidade, pode ser expressa com distúrbios do ritmo, disfunção ventricular e envolvimento valvular, que deixou a sua evolução natural pode levar à morte do paciente, dando relevância para o reconhecimento precoce pelos pro-fissionais da etiologia hormonal de IC e/ou a causa de sua descompensação. Este diagnóstico permite o tratamento precoce e eficaz como os estados de eutireoidismo conduzir à normalização da doença cardiovascular na maioria dos pacientes.
Palavras-chave: Hipertireoidismo; Hormônio da tireóide; Sistema cardiovascular; Fibrilação atrial; Insuficiência cardíaca; Lesão miocárdica; Hipertensão pulmonar
Abreviaturas
CV: cardiovascular.
HT: hormona tiroidea.
ICC: insuficiencia cardíaca crónica.
IC: insuficiencia cardíaca.
T3: triyodotironina.
TSH: tirotrofina.
T4: tiroxina.
ADN: ácido desoxirribonucleico.
RT: receptores nucleares de las hormonas tiroideas.
SERCA2: retículo sarco/endoplasmático.
CPM: cadenas pesadas de miosina.
ATPasa: adenosina trifosfatasa.
VI: ventrículo izquierdo.
TRAB: anticuerpos antirreceptores de TSH.
GC: gasto cardíaco.
FE: fracción de eyección.
HVI: hipertrofia del ventrículo izquierdo.
FC: frecuencia cardíaca.
RVS: resistencia vascular sistémica.
PET: tomografía por emisión de positrones.
FA: fibrilación auricular.
ESV: extrasístoles supraventriculares.
R3: tercer ruido cardíaco.
HTP: hipertensión pulmonar.
BB: betabloqueantes.
EPOC: enfermedad pulmonar obstructiva crónica.
MMI: metilmercaptoimidazol.
VD: ventrículo izquierdo.
L-NAME: nitro-L-arginina metil éster.
IMT: íntima media.
IAM: infarto agudo de miocárdio.
ACV: accidente cerebrovascular.
Introducción
Desde hace más de 200 años, se reconoce la relación existente entre la hormona tiroidea (HT) y el sistema cardiovascular (CV). La glándula tiroides y el corazón están estrechamente relacionados desde el punto de vista embriológico. Esta relación fisiológica se ve reforzada por los predecibles cambios de la función cardiovascular en los distintos trastornos tiroideos. De hecho, las manifestaciones cardiovasculares están entre las más frecuentes y típicas presentaciones del hipertiroidismo1.
Para entender, diagnosticar y tratar las cardiopatías que acompañan al hipertiroidismo, es importante conocer bien los mecanismos celulares de la acción de las hormonas tiroideas sobre el corazón y sobre las células del músculo liso2. El hipertiroidismo no solamente puede agravar una enfermedad cardíaca preexistente, sino que también puede conducir a una enfermedad cardíaca.
Son los objetivos de esta revisión: 1) profundizar en el conocimiento de los mecanismos de acción de la HT sobre el corazón y sistema vascular periférico, y los cambios anátomo-estructurales que se producen a nivel miocárdico; 2) enfatizar en las primeras manifestaciones cardíacas de la enfermedad, impidiendo su evolución natural hacia la insuficiencia cardíaca crónica (ICC), dando síntomas iniciales como palpitaciones hasta avanzar a un estado de mayor complejidad como es la insuficiencia cardíaca (IC), entidad de alta morbimortalidad, pudiéndose presentar con alteraciones del ritmo, deterioro en la función ventricular y afectación valvular; ellos dejados a su curso natural pueden llevar a la muerte del paciente; 3) dar relevancia a la utilidad de un diagnóstico temprano por parte del profesional, en la probable etiología hormonal de la IC como causa de descompensación, para realizar un tratamiento precoz, siendo que estados de eutiroidismo llevan a la normalización de los trastornos cardiovasculares en la mayoría de los pacientes.
Mecanismos celulares y moleculares del efecto de la hormona tiroidea sobre el corazón
La HT tiene relevantes acciones sobre el corazón y la circulación, generando múltiples cambios que incluyen alteraciones hemodinámicas y efectos mediados sobre el miocito cardíaco a través de la expresión génica. En 1786, Parry demostró las características clínicas de la tirotoxicosis: palpitaciones, pulso irregular y disnea. En 1835, Graves aportó una descripción "bocio-tóxica". Las manifestaciones cardíacas de la tirotoxicosis condujeron a conclusiones erróneas sobre que la enfermedad se originaba dentro del corazón. En 1918, Zondek describió a un paciente con las características del corazón mixedematoso: silueta cardíaca dilatada, bajo voltaje cardíaco y acción cardíaca enlentecida. La triyodotironina (T3) fue descubierta por Pitt-Rivers y Gross en el año 1952, y su producción endógena fue descripta por Ingbar, Sterling y Braverman en 1970. Condliffe, en el año 1963, aisló la tirotrofina (TSH). En el año 1971 Mayberry y Hershman describieron, simultáneamente, el test de inmunoensayo de la TSH para el diagnóstico de hipotiroidismo3. A pesar de las asociaciones precoces entre el sistema CV y enfermedad tiroidea, es sólo reciente el hecho de considerar a la HT como un agente terapéutico potencial en la enfermedad CV. Para comprender las alteraciones en la función cardíaca que acompañan al hipertiroidismo es necesario reconocer los mecanismos a través de los cuales la HT actúa en el miocito cardíaco y en las células del músculo liso vascular.
La síntesis de tiroxina (T4) y de T3 sucede dentro de la glándula tiroides. La T4 es el producto principal mayoritariamente inactivo. La conversión de T4 a T3 no ocurre en el miocito. El 85% de T3, el componente biológicamente activo, es derivado de la conversión periférica de T4 por la enzima 5 monodeiodinasa principalmente ocurre en el hígado y riñón (Figura 1). Tanto la T4 como la T3 circulan casi enteramente (95%) unida a la familia de proteínas y el 5% restante lo hace libremente. La T3 es la HT biológicamente relevante en el miocito cardíaco, así como en otras células, hay evidencia de que las membranas celulares contienen proteínas transportadoras específicas para T3. Diversos estudios han confirmado que T3 (forma activa de la HT) explica la inmensa mayoría de sus efectos biológicos (Figura 1), entre ellos: la estimulación de la termogénesis tisular, las alteraciones en la expresión de diversas proteínas celulares, y los efectos sobre el corazón y las células musculares lisas1,4. La T3 libre sérica entra en las células mediante un proceso de difusión facilitada y parece pasar directamente al núcleo sin unirse a otra proteína dentro de la célula (Figura 2). La mayoría de las observaciones indican que los miocitos cardíacos no pueden metabolizar T4 ni T3 y, por lo tanto, todos los efectos nucleares y los cambios de expresión génica se deben a los cambios en las concentraciones sanguíneas de T3.
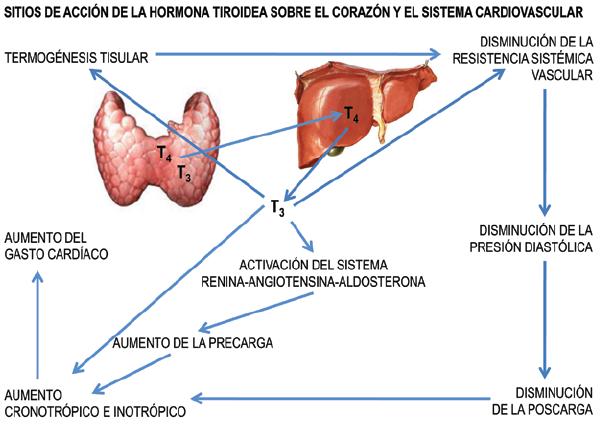
Figura 1. Esquema del metabolismo de la hormona tiroidea y de los efectos de triyodotironina sobre el corazón y los vasos sistémicos.
T4: tiroxina. T3: triyodotiroxina.
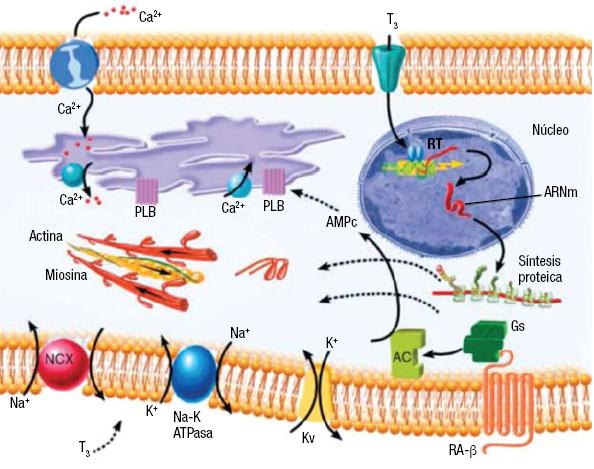
Figura 2. Triyodotironina (T3) entra en la célula y se une a receptores nucleares T3. El complejo se une a elementos sensibles a hormona tiroidea (EST) y regula la transcripción de determinados genes. Se representan también las acciones no nucleares de T3 sobre los canales iónicos de sodio, potasio y calcio. AC: adenilciclasa. AMPc: adenosina monofosfato cíclico. ARNm: ARN mensajero. ATPasa: adenosina trifosfato. Kv: canal de potasio activado por voltaje. NCX: canal de sodio. PLB: fosfolambano. RA-β: receptor adrenérgico beta. RT: proteína receptora de T3.
Una vez que alcanza el miocito, interacciona con moléculas fuertemente asociadas a la cromatina conocidas como"receptores nucleares de las hormonas tiroideas (RT)". Los RT3 pertenecen a las "superfamilias de receptores nucleares", los cuales derivan evolutivamente de un gen ancestral común. Cada uno de ellos es un factor de transcripción nuclear dependiente del ligando que regula la velocidad de transcripción de genes blanco por medio de una unión de secuencias específicas de ácido desoxirribonucleico (ADN), generalmente, ubicadas en la región 5´-flanqueante de estos genes. Los receptores nucleares se unen al ADN como monómeros, aunque la mayoría lo hace como homo o heterodímeros, compuesto por receptores nucleares de T3, y otro receptor de la familia de los receptores de hormonas esteroideas. La unión de estos receptores con T3, en combinación con otros coactivadores, conduce a una activación transcripcional óptima. Ante la ausencia de T3, los receptores inhiben genes que son estimulados con la HT. Es importante destacar que la forma "heterodimérica" es transcripcionalmente más activa en el caso de RT como así también de otros receptores nucleares de hormonas no esteroideas.
Los miocitos cardíacos expresan las isoformas alfa y beta del receptor de hormona tiroidea (RTalfa 1 y RTalfa 2), que proceden de dos genes distintos (Figura 3). Estos genes dan origen por acoplamiento, a las variantes RTalfa 1 y RTalfa 2, de las cuales sólo la primera se une a la HT, así como a RTbeta 1, RTbeta 2 y RTbeta 34. Se ha propuesto, aunque sin pruebas directas que lo confirmen, que RTalfa 1 es la principal isoforma fijadora de T3 en el corazón y que la isoforma específica podría determinar, a su vez, la expresión génica específica del miocito4,5. De modo similar a las familias de proteínas receptores de esteroides y ácido retinoico, los RT actúan uniéndose en forma de homodímeros o heterodímeros a los elementos sensibles a la hormona tiroidea de la región promotora de determinados genes. La unión a regiones promotoras podría activar o reprimir la expresión del gen6.

Figura 3. Genes de la cadena pesada de miosina cardíaca, que muestran su localización uno tras otro en el brazo corto del cromosoma 17, en seres humanos, y las proteínas muy similares que son producidas168.
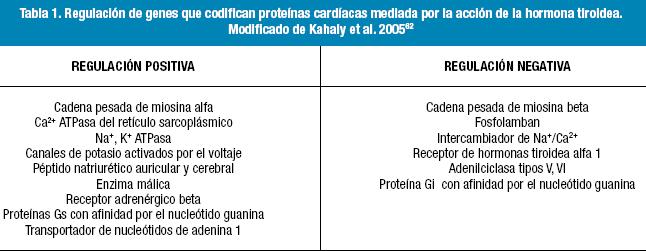
Las proteínas cardíacas cuya transcripción es regulada por la HT se enumeran en la Tabla 1. Entre ellas hay proteínas estructurales y reguladoras relacionadas con la función contráctil, incluyendo el calcio ATPasa del retículo sarco/endoplásmico (SERCA2), fosfolamban y las cadenas pesadas de miosina (CPM)7, así como una amplia variedad de canales iónicos de membrana cardíacos y receptores de la superficie celular, lo cual proporciona un mecanismo molecular para explicar muchos de los efectos de la HT en el corazón. Las primeras proteínas descriptas y la mejor estudiada hasta la fecha han sido las isoformas alfa (estimulada por la HT) y beta (inhibida por la HT) de la cadena pesada de miosina8 (proteínas miofibrilares que componen el filamento grueso del aparato contráctil del miocito cardíaco) (Figura 3). Sin embargo, en el ventrículo humano, la principal isoforma de miosina expresada es la beta y esto no parece modificarse apenas en las diversas enfermedades tiroideas9, cambios de insuficiente magnitud para considerar cambios funcionales. Se observan cambios de expresión de isoforma de la cadena pesada de miosina, en aurículas humanas en diversas enfermedades como la insuficiencia cardíaca congestiva, y no se ha comprobado todavía, si estos cambios son mediados por la hormona tiroidea10.
La adenosina trifosfatasa (ATPasa) activada por calcio del retículo sarcoplásmico es una importante bomba iónica, que determina la magnitud de los movimientos de calcio en el miocito. La recaptación de calcio hacia el retículo sarcoplásmico al comienzo de la diástole determina, en parte, la velocidad de relajación del ventrículo izquierdo (VI) (fase de relajación isovolumétrica)2. La actividad de la calcio ATPasa del SERCA2, a su vez, está regulada por la proteína polimérica fosfolambano, cuya acción capaz de inhibir la actividad de SERCA depende del nivel de fosforilación de los monómeros de fosfolamban11, Los fármacos inotrópicos que estimulan la contractilidad cardíaca a través del aumento del AMPc (adenosina monofosfato cíclico) en el miocito, lo hacen estimulando la fosforilación de fosfolamban. La HT inhibe la expresión genética de fosfolamban y aumenta su fosforilación12, y animales modificados genéticamente con deficiencia de fosfolamban, no presentan aumento de la contractilidad cardíaca cuando son expuestos a un exceso de HT11. Todas estas observaciones indican que la hormona tiroidea ejerce la mayor parte de sus efectos directos sobre la contractilidad cardíaca a través de la regulación de los movimientos de calcio mediante el sistema SERCA-fosfolambano, tanto en la transcripción como después de ésta. Este mecanismo molecular podría explicar por qué la función diastólica varía inversamente con el espectro de enfermedades tiroideas (Figura 4)2,13,14. Además en el hipertiroidismo, el bloqueo betaadrenérgico cardíaco no reduce la relajación diastólica rápida, esto separa aún más a la HT de los efectos adrenérgicos del hipertiroidismo2.
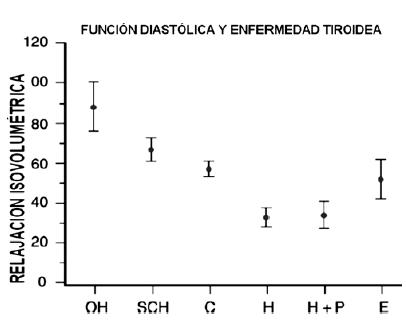
Figura 4. La función diastólica, medida por el tiempo de relajación isovolumétrica, varía a lo largo del espectro de tiroidopatías, desde el hipotiroidismo clínico (OH), hipotiroidisomo subclínico (SCH), normales (C), hipertiroidismo (H), hipertiroidismo tras bloqueo adrenérgico (H+P) e hipertiroidismo tratado para restablecer la función tiroidea normal (E)44.
Cambios en otros genes del miocito, entre ellos el de la Na+/K+ ATPasa explican el aumento del consumo basal de oxígeno del corazón en el hipertiroidismo experimental, así como la disminución de la sensibilidad a la digital de los pacientes hipertiroideos. Diversos estudios han demostrado que la HT puede regular la expresión génica de sus propios receptores nucleares dentro de los miocitos cardíacos (Tabla 1) (receptores beta-adrenérgicos) y otras proteínas reguladoras como la guanina-nucleótido y adenilciclasa tipo V y VI. La estimulación de receptores beta-adrenérgicos causa un aumento intracelular de segundos mensajeros (AMPc) que conduce a una despolarización diastólica acelerada y aumento de la frecuencia cardíaca.
Varios transportadores iónicos, tales como Na+/K+ ATPasa, intercambiador de Na+/Ca2+ y los canales de potasio dependientes de voltaje, son también regulados por los niveles de HT, los cuales coordinan la respuesta electroquímica y mecánica del miocardio2.
Además de los conocidos efectos nucleares de la HT, cada vez se descubren más respuestas cardíacas a la HT que parecen estar mediadas por mecanismos no genómicos15, tal y como lo indican su relativamente rápido comienzo de acción (tan rápido que no puede explicarse por cambios en la expresión de genes y en la síntesis de proteínas) y la falta de sensibilidad a los inhibidores de la transcripción génica. No se ha determinado la importancia de estas acciones. Podrían alterar las propiedades funcionales de canales iónicos de la membrana como el canal de sodio y la corriente rectificadora entrante de potasio.
Pruebas de función tiroidea
Una serie de pruebas de laboratorio sensibles y específicas pueden establecer el diagnóstico de las tiroidopatías con gran precisión. La más utilizada es la TSH sérica, que es la determinación más sensible para el diagnóstico del hipertiroidismo primario16.
El hipertiroidismo puede ser transitorio o persistente. El persistente se puede dividir en causas endógenas, enfermedad de Graves, nódulo autónomo y bocio multinodular, y causas exógenas: sobretratamiento con HT. En el caso del transitorio se debe a una tiroiditis.
Las concentraciones séricas de TSH debido a la autorregulación normal por las concentraciones elevadas de T4 (y T3) sobre la síntesis y secreción hipofisiaria de TSH están bajas (≤0,1 μUI/ml). Las determinaciones de T4 libre y de T3 libre pueden ser útiles cuando las concentraciones de globulina transportadora de tiroxina pueden estar alteradas por trastornos hepáticos, nutricionales, genéticos concomitantes y embarazo (Tabla 2).

Las tiroidopatías autoinmunitaria (Hashimoto y Graves) pueden diagnosticarse con ayuda de determinaciones serológicas de anticuerpos antitiroideos, especialmente de anticuerpos antitiroperoxidasa y antitiroglobulina, anticuerpos antirreceptores de TSH (TRAB). La curva de captación de yodo131 es una prueba funcional de utilidad para confirmar el hipertiroidismo. La captación a las 24 horas y el centellograma con I131 o tecnecio99, son pruebas útiles para el diagnóstico, si el hipertiroidismo está producido por una tiroiditis: hashi-tirotoxicosis en la etapa destructiva, la captación será cercana a cero, y si el hipertiroidismo está producido por una enfermedad de Graves, la captación será elevada y con una imagen centellográfica homogénea.
Los adenomas autónomos muestran una imagen de nódulo caliente hiperfuncionante, y el bocio multinodular tóxico muestra un patrón moteado o heterogéneo. La guía de la American Thyroid Association17 recomienda que en presencia de TSH baja se debe realizar el estudio centellográfico junto a la ecografía para evaluar el comportamiento de los nódulos y si son mayores de 1 a 1,5 cm realizar el estudio citológico.
Interacción de la HT con el sistema nervioso simpático (catecolaminas)
Las primeras observaciones sobre el corazón en el hipertiroidismo, se pusieron de manifiesto en su similitud, con los estados hiperadrenérgicos, e incluso sugieren la existencia de hipersensibilidad a las catecolaminas. En este postulado se basa la prueba descripta por Goetsch en 1918, que diagnosticaba el hipertiroidismo demostrando una respuesta exacerbada de cardioaceleración y aumento de la presión arterial ante dosis subcutáneas de adrenalina.
La determinación de las concentraciones de catecolaminas circulantes en los sujetos hipertiroideos puso de manifiesto que, a pesar de la aparente exacerbación de los signos y síntomas adrenérgicos, las concentraciones de adrenalina y noradrenalina estaban disminuidas. Este descubrimiento originó el concepto de hipersensibilidad a las catecolaminas, que fue apoyado por la demostración molecular de un aumento de receptores adrenérgicos β1 en los miocitos cardíacos en el hipertiroidismo experimental. Un estudio perfectamente controlado, efectuado con primates, sin embargo, ha demostrado claramente que no existe hipersensibilidad a las catecolaminas en el corazón o el aparato cardiovascular en el hipertiroidismo experimental18. Aparte de aumentar la cantidad de receptores adrenérgicos β1 y de proteínas transportadoras de guanosina trifosfato, la HT reduce la expresión génica de isoformas de la subunidad catalítica de adenilciclasas específicas del corazón (V, VI) y, por tanto, mantiene la respuesta celular a los betabloqueantes dentro de los límites normales19. El tratamiento con betabloqueantes mejora los signos y síntomas cardiovasculares asociados con el hipertiroidismo20, la frecuencia cardíaca es disminuida, pero el aumento en el rendimiento diastólico no se altera con el tratamiento, lo que indica la acción directa de la HT sobre el corazón y el aumento de calcio21.
Alteraciones hemodinámicas en el hipertiroidismo
Contractilidad miocárdica
El término "contractilidad miocárdica" se refiere a la propiedad intrínseca del músculo cardíaco para generar trabajo22, por lo tanto esto corresponde al rendimiento del corazón independientemente del efecto de la frecuencia cardíaca y condiciones de carga (precarga/poscarga). En contraste, "función sistólica ventricular" representa el efecto agregado de todos los mecanismos que controlan el rendimiento cardíaco (frecuencia cardíaca, precarga, poscarga y contractilidad miocárdica).
En pacientes hipertiroideos, hay una consistente mejoría de la función sistólica del VI en reposo23. Hay dos escuelas de pensamiento acerca de cómo debe ser interpretada la contractilidad miocárdica. La controversia está dada por los cambios en la frecuencia cardíaca y las condiciones de carga.
Merillon y colaboradores24 sugieren que el alto gasto cardíaco (GC) es probablemente debido a la interacción sinérgica entre el aumento de la frecuencia cardíaca y la precarga ventricular. Por el contrario Feldman y col.25 encontraron que la velocidad de acortamiento en las fibras miocárdicas fue más alta en pacientes con hipertiroidismo, independientemente de la precarga y frecuencia cardíaca. Este hallazgo provee evidencia adicional de una acción directa inotrópica de la HT, cambios en la fracción de eyección (FE) que no estaban asociados a las condiciones de carga ventriculares. Sin embargo, la reserva contráctil puede estar disminuida en pacientes con hipertiroidismo, este concepto emerge de la observación que la FE ventricular, aunque está elevada en el reposo, no aumenta en el ejercicio26.
El efecto directo de la HT para producir aumento del inotropismo cardíaco se debe a múltiples mecanismos:
- Estimula los receptores beta, incrementando los niveles de AMPc intracelular y elevando los niveles de calcio y la densidad de los canales de calcio tipo L.
- Aumenta la expresión de Ca2+ ATPasa del retículo sarcoplásmico.
- Modifica los factores hemodinámicos, ya sea alterando la poscarga, precarga o la frecuencia cardíaca.
- Promueve la expresión de miocitos de la isoforma de cadena pesada de miosina B, aumentando la expresión de Ca2+ ATPasa del retículo sarcoplásmico y disminuyendo la expresión de la Ca2+ ATPasa sarcolémica que regula a la fosfolamban.
- Modula la expresión de la Na+/K+ ATPasa2.
Función ventricular izquierda
En el hipertiroidismo subclínico se observa una disfunción cardiovascular, especialmente, una disfunción diastólica27-32.
El incremento en los valores de la hormona tiroidea produce un aumento en la contractilidad, relajación, volumen minuto y de la frecuencia cardíaca33,34. Estos cambios pueden incrementar el daño miocárdico y subsecuentemente desarrollar hipertrofia del ventrículo izquierdo (HVI) y disfunción cardíaca.
Al realizar un ecocardiograma Doppler en los pacientes con hipertiroidismo subclínico se encuentra un incremento en los valores del Doppler tisular pulsado. Aquellos pacientes con valores inferiores de TSH a 0,50 µUI/ml, que presentaban valores elevados de Doppler tisular, fueron concordantes con los estudios previos en pacientes hipertiroideos35-37. Por ende, el hipertiroidismo subclínico presenta una acción a nivel cardíaca detectable por el Doppler pulsado tisular, pero aún no se sabe su relevancia clínica.
Esta medición, sumada a la ecocardiografia convencional, es una buena herramienta para detectar la disfunción diastólica, con una mejor sensibilidad y especificidad, comparado a la medición del Doppler mitral en forma aislada. Es importante la búsqueda de la disfunción diastólica debido a que la misma se relaciona con un aumento en la morbi-mortalidad.
Se deberán realizar mayores estudios con Doppler pulsado, en los pacientes con hipertiroidismo subclínico, no sólo para valorar la posible progresión de la disfunción diastólica sino también para valorar si el tratamiento puede generar la regresión de la misma.
Propiedades diastólicas del ventrículo izquierdo
El llenado del VI puede estar influenciado por su hipertrofia, relajación diastólica y la precarga, propiedades que pueden ser alteradas por la HT.
La hipertrofia del VI en el hipertiroidismo puede estar dada por la estimulación en la síntesis de proteínas miocárdicas o el aumento en la precarga38.
La función ventricular diastólica ha sido valorada en pacientes con hipertiroidismo, pacientes que fueron estudiados en el diagnóstico inicial, 2 semanas posteriores al tratamiento con propanolol y 4-6 meses en estado de eutiroidismo. Aquellos tratados con propanolol disminuyeron la frecuencia cardíaca a valores normales, pero no disminuyó el tiempo de relajación isovolumétrica que permaneció marcadamente acelerado39. Estos hallazgos son consistentes con el concepto que la HT aumenta la función diastólica del VI, independientemente, de los mecanismos adrenérgicos y de la frecuencia cardíaca. Esto se halla asociado al aumento de la actividad del SERCA240.
Sin embargo, en pacientes con hipertiroidismo e hipertrofia ventricular, el llenado diastólico puede estar perjudicado porque los efectos beneficiosos en la mejoría de la relajación son contrarestados por el aumento en la rigidez ventricular.
Efecto cronotrópico
La frecuencia cardíaca (FC) es un importante mecanismo para regular el gasto cardíaco. Además de determinar la tasa de eyección cardíaca, afecta a la función sistólica y diastólica. Una FC acelerada aumenta el GC a cualquier nivel dado de precarga, un hallazgo compatible con la mejoría de la contractilidad miocárdica20. Una FC elevada también aumenta el porcentaje de relajación miocárdica, mejorando el llenado rápido ventricular (efecto lusotrópico)20.
Sin embargo, la FC acelerada no aumenta el rendimiento cardíaco si la precarga no está aumentada o al menos, se mantiene constante. El ritmo inducido aumenta la frecuencia de contracción y generalmente reduce la precarga y volumen de eyección, así el GC permanece constante41.
Por otra parte, un aumento de la FC reduce el tiempo de llenado diastólico y, por lo tanto, conduce a una dependencia de la sístole auricular. Esto explica el importante impacto fisiopatológico de la fibrilación auricular (FA) en el rendimiento cardíaco.
Estudios del ritmo auricular en sujetos normales han demostrado que un aumento de la FC reduce la dinámica adaptación del árbol arterial y aumenta la presión arterial42. Este efecto probablemente resulte por el tiempo alterado de la presión de onda reflejada que retorna del árbol arterial periférico consecuencia de la reducción en la duración absoluta de la sístole. Es decir, como el tiempo sistólico disminuye porque está aumentada la FC, la onda de presión reflejada que retorna del árbol arterial periférico se agregaría y aumentaría la presión de onda que va hacia delante, con lo cual aumentaría la presión arterial (Figura 5).

Figura 5. Efecto potencial del aumento de la frecuencia cardíaca sobre la presión arterial en pacientes con hipertiroidismo49.
Otros conceptos que sugieren el efecto directo cronotrópico de la HT sobre el corazón son:
- En estudios in vitro en fibras auriculares y nodo sinusal de conejos, se encontró disminución en la duración de la fase de repolarización del potencial de acción y aumento de la despolarización diastólica y por lo tanto la razón de contracción43.
- Estudios en corazones aislados de animales, con inducción de hipertiroidismo, mostraron aumento en la frecuencia cardíaca con períodos refractarios acortados más que en animales eutiroideos44.
Los mecanismos por el cual la HT induce estos cambios electrofisiológicos permanecen todavía desconocidos, pero pueden estar relacionados en parte por sus efectos sobre la densidad de bomba de Na+ y aumento de la permeabilidad al Na+ y al K+.
Precarga
La precarga es la fuerza hemodinámica ejercida sobre la pared ventricular durante el llenado y corresponde a la tensión o estrés de la pared ventricular al final de la diástole. La precarga juega un rol importante en regular la fracción de eyección del corazón (mecanismo de Frank-Starling). De hecho, es el mecanismo más eficiente por el cual el GC es ajustado a la demanda metabólica periférica. En un organismo intacto, la precarga está regulada en gran medida por el retorno venoso, que a su vez depende de la resistencia vascular sistémica y del tono venoso. El volumen total de sangre y la contracción auricular pueden contribuir también significativamente en la regulación de la precarga45.
Aún no se ha establecido si el impacto del hipertiroidismo sobre la precarga cardíaca en humanos contribuye al aumento del rendimiento ventricular izquierdo. Gibson y Harris46 y Anthonisen y col.47 demostraron que el volumen sanguíneo es aumentado en pacientes hipertiroideos, lo que apoya que la precarga tiene un rol predominante en determinar un alto GC. En forma similar, Resnick y Laragh48 demostraron que el sistema renina-angiotensina-aldosterona está activado en el hipertiroidismo. La HT ha mostrado un "up-regulation" en la secreción de eritropoyetina, con un aumento en la cantidad de glóbulos rojos, lo que también contribuye a un aumento en el volumen sanguíneo y la precarga49,50. Otra evidencia está provista por el aumento del llenado temprano del VI y por su rápida relajación, independientemente, de la FC y de las catecolaminas26. De hecho, el aumento del índice de la velocidad del flujo temprano transmitral y el acortamiento del tiempo de relajación isovolumétrica pueden reflejar el retorno venoso aumentado, el cual conduce a un aumento del gradiente de presión transmitral en la protodiástole (debido al aumento de la presión auricular) y la apertura temprana de la válvula mitral21. Alternativamente, el tiempo acortado de la relajación isovolumétrica puede ser debido a la mejoría de la función diastólica, la cual a su vez permitiría el aumento del retorno venoso sin cambios relevantes en la presión de llenado. Al acentuarse el período isovolumétrico decae la presión intracavitaria, y el aumento del índice de relajación aumenta la succión ventricular51. En consecuencia, el mayor gradiente de presión temprano aurículo-ventricular estaría dado por un aumento de la succión ventricular más que por un aumento en la presión auricular22 (Figura 6).

Figura 6. Efecto de succión atrio (línea cortada) ventricular (línea sólida) sobre el gradiente de presión y la velocidad de flujo Doppler transmitral49.
Poscarga y regulación de la presión arterial
La poscarga es la fuerza hemodinámica ejercida sobre la pared ventricular durante la eyección, correspondiendo por lo tanto, a la presión de fin de sístole. Esta contribuye en determinar el volumen de fin de sístole ventricular y modular el rendimiento miocárdico. Por lo tanto, la poscarga es un determinante de la fracción de eyección del corazón, proporcionando un mecanismo crucial de acoplamiento de la función de bomba al sistema arterial. En organismos intactos, la poscarga está regulada por la presión arterial (poscarga vascular); la cual, a su vez, depende de la interacción entre la resistencia vascular y componentes pulsátiles de la carga arterial (impedancia aórtica, relajación arterial e índice de propagación y reflexión de la onda arterial).
Cambios en el tamaño y la geometría ventriculares pueden afectar la poscarga ventricular41. En muchos artículos de revisión sobre los efectos de la hormona tiroidea en el sistema cardiovascular, se reporta que la poscarga ventricular se encuentra disminuida en pacientes hipertiroideos52. Esta idea ganó credibilidad gracias a la conclusión de que la HT promueve directamente la relajación del músculo liso53. Sin embargo, la reducción en la carga arterial no está acompañada por la reducción del estrés parietal al final de sístole del VI en pacientes hipertiroideos, en gran medida porque hay una tendencia hacia una presión arterial elevada54. Esta observación apoya firmemente la hipótesis de que en el hipertiroidismo, a pesar de una marcada reducción en la resistencia vascular, la carga del pulso arterial sufre un cambio compensatorio. Como resultado de ello, por mantener e incluso aumentar la impedancia aórtica, la carga arterial pulsátil sostiene la presión sistólica arterial y previene la reducción de la poscarga ventricular. El mecanismo subyacente que aumenta los componentes pulsátiles de la poscarga vascular en el hipertiroidismo no está del todo claro. Aunque se especula que el aumento de la frecuencia cardíaca en el hipertiroidismo podría jugar un rol importante en este mecanismo, porque ésta reduciría la adaptación del árbol arterial, por lo que aumentaría la impedancia aórtica y, a su vez, aumentaría la presión arterial sistólica55.
Efectos en la circulación periférica
Los efectos directos sobre las células musculares lisas reducen la resistencia vascular sistémica (RVS) de las arteriolas de la circulación periférica. La HT al disminuir la RVS hace caer el volumen efectivo arterial, causando un aumento en la actividad del sistema renina-angiotensina y una estimulación del eje angiotensinógeno-aldosterona, generando una mayor absorción de sodio renal, que conduce a un aumento del volumen plasmático. La HT estimula la secreción de eritropoyetina. La combinación de estas dos acciones incrementa el volumen sanguíneo y la precarga cardíaca49. Por lo tanto, la disminución de la RVS (de hasta un 50%) junto con el aumento del retorno venoso y de la precarga incrementa el GC, pudiendo duplicarlo. La combinación de un volumen sanguíneo aumentado y una mejoría de la relajación diastólica del corazón contribuyen a un aumento del volumen de fin de diástole del VI o precarga; en forma similar, la caída de las resistencias vasculares periféricas y la mejoría de la contractilidad miocárdica determinan un menor volumen de fin de sístole ventricular izquierdo o poscarga. El efecto neto de estos 2 hechos es un aumento del volumen de eyección ventricular izquierdo.
La HT aumenta el consumo de oxígeno y los requerimientos a nivel periférico, lo que ocasiona secundariamente un aumento en la contractilidad miocárdica, sumado a su efecto directo. La vasodilatación es debida a una acción directa de T3 sobre las células musculares lisas y a través de su acción indirecta por la liberación de vasodilatadores locales, secundariamente a su actividad metabólica y de consumo de oxígeno. En cuanto a su acción directa, T3 modifica los canales de sodio y potasio, produciendo una disminución de la contractilidad del músculo liso y del tono vascular.
Estudios sobre el metabolismo del acetato, realizados mediante tomografía por emisión de positrones (PET), han demostrado que el gran aumento del GC observado en el hipertiroidismo se consigue sin modificaciones de la eficacia energética56. La HT parece reducir la RVS a través de efectos directos sobre las células musculares lisas y de cambios en el endotelio vascular, en los cuales puede estar implicada la síntesis y secreción de óxido nítrico. El efecto vasodilatador de T3 puede observarse unas horas después de la administración de la misma a pacientes sometidos a injerto de derivación arterial coronaria y a pacientes con insuficiencia cardíaca congestiva9,57. La importante contribución en la disminución de la RVS y al aumento del flujo sanguíneo en pacientes con hipertiroidismo es evidenciado por los resultados de estudios en los cuales la administración de vasoconstrictores arteriales, atropina o fenilefrina, disminuyen el flujo de sangre periférico y el GC hasta en un 34%, pero no tienen efecto en sujetos normales58.
Así pues, la combinación del incremento del GC y la disminución de la distensibilidad arterial, que puede ser más acusada en pacientes ancianos con cierto grado de arteriopatía, provoca hipertensión hasta en un 30% de los pacientes59.
En la Tabla 3, se comparan los cambios hemodinámicos que se producen a nivel cardiovascular en pacientes con hipertiroidismo contra valores normales de la HT2 y en la Figura 7 se grafican a modo de resumen los mecanismos fisiopatológicos por los que aumenta el GC en pacientes con hipertiroidismo.

Figura 7. Mecanismo fisiopatológico que demuestra que el gasto cardíaco en reposo aumenta en pacientes con hipertiroidismo49. RVS: resistencia vascular sistémica. SRAA: sistema renina-angiotensina-aldosterona. PAD: presión arterial diastólica. PAS: presión arterial sistólica. PAM: presión arterial media. VFDVI: volumen de fin de diástole del ventrículo izquierdo. VFSVI: volumen de fin de sístole del ventrículo izquierdo. PP: presión de pulso.
Manifestaciones clínicas del hipertiroidismo: generales y a nivel cardiovascular
Clásicamente, los pacientes con hipertiroidismo manifiestan diversos signos y síntomas de acuerdo a los diferentes sitios de acción que tiene la HT, algunos de ellos son: nerviosismo, labilidad emocional, temblor, reflejos vivos (sistema nervioso); pérdida de peso con igual ingesta, hiperdefecación (aparato gastrointestinal); debilidad, atrofia muscular, osteopenia (resorción ósea por acción directa de las HT sobre el osteoclasto); piel caliente, húmeda, suave, palmas húmedas y calientes, onicólisis (piel y anexos); oligomenorrea, alteración de la fertilidad, ginecomastia, impotencia (aparato reproductor); pérdida de peso, fatiga, sudoración aumentada, intolerancia al calor (metabolismo)60.
Los síntomas cardiovasculares representan a menudo la forma predominante de la manifestación de los pacientes con hipertiroidismo. La mayoría tiene palpitaciones provocadas por el aumento de la FC y de la fuerza de la contractilidad cardíaca61. El aumento de la FC se debe al aumento del tono simpático y de la disminución de la estimulación parasimpática60. Es frecuente encontrar una FC superior a 90 latidos/minuto, tanto en reposo como durante el sueño; la variación diurna normal de la FC está reducida y existe un aumento exagerado durante el ejercicio. Muchos enfermos hipertiroideos presentan intolerancia al ejercicio y disnea de esfuerzo, en parte, debido a la debilidad de los músculos esqueléticos y respiratorios. La reserva funcional cardíaca se ve comprometida en caso de resistencias vasculares bajas y aumento de la precarga, y no puede aumentar para adecuarse a las demandas del ejercicio máximo o submáximo (intolerancia al ejercicio)62.
Algunos pacientes hipertiroideos pueden experimentar dolor torácico de tipo anginoso. En pacientes ancianos con enfermedad arterial coronaria conocida o sospechada, el aumento del trabajo cardíaco provocado por el aumento del GC y de la contractilidad cardíaca puede producir isquemia cardíaca (disbalance entre la oferta y la demanda), que puede responder con betabloqueantes o al restablecimiento del estado eutiroideo63. En un número pequeño de pacientes, generalmente mujeres jóvenes, es posible la aparición de un síndrome caracterizado por dolor torácico en reposo asociado a cambios isquémicos en el electrocardiograma. El cateterismo cardíaco ha demostrado que la mayoría de estos pacientes presentan arterias angiográficamente normales, observándose vasoespasmo coronario similar al de la angina variante64. El tratamiento satisfactorio del hipertiroidismo revierte estos síntomas65.
Con menor frecuencia se puede hallar un ruido sistólico sobre le segundo espacio intercostal izquierdo (roce de Means-Lerman) con algunas características auscultatorias del roce pericárdico imitando una pericarditis66. Otros conjunto de síntomas y signos cardiovasculares se presentan marcando un grado mayor de compromiso orgánico, como son la insuficiencia cardíaca y las arritmias (taquicardia sinusal, fibrilación auricular, etc.).
Trastornos en la actividad eléctrica
Hormona tiroidea y efectos electrofisiológicos
La HT ejerce una marcada influencia sobre la generación (efecto cronotrópico) y conducción (efecto dromotrópico) del impulso eléctrico. La triyodotironina aumenta la tasa de despolarización sistólica y la de repolarización diastólica, disminuyendo la duración del potencial de acción y del período refractario del miocardio auricular, tanto como el período refractario del nodo aurículo-ventricular. El mecanismo por el cual la T3 induce cambios electrofisiológicos está relacionado en parte por su efecto sobre el aumento de la densidad y permeabilidad de las bombas de Na+/K+.
Hipertiroidismo y fibrilación auricular
La HT tiene una importante influencia sobre la función del nodo sinusal, actuando en forma directa y produciendo aumento en su actividad intrínseca y, consecuentemente, aumentando la FC sin mediar el sistema nervioso autónomo67.
La taquicardia sinusal es la alteración más común y se encuentra en casi todos los pacientes con hipertiroidismo68. Un aumento de la FC en reposo es característico de esta enfermedad. Sin embargo, la FA es más comúnmente identificada con el hipertiroidismo69, arritmia que está asociada per se a un aumento en la morbilidad y mortalidad cardiovascular70,71. La prevalencia de la FA en esta enfermedad se encuentra entre el 2% y el 20%. En comparación con una prevalencia de FA del 2,3% en una población con función tiroidea normal, la prevalencia del FA en pacientes con hipertiroidismo clínico es del 13,8%72. La FA en pacientes con hipertiroidismo se incrementa con la edad, por década, con un máximo del 15% en los pacientes mayores de 70 años73.
Ocurre más frecuentemente en hombres que en mujeres, aunque la incidencia de hipertiroidismo es 5 a 10 veces mayor en mujeres que en los hombres. La severidad de la tirotoxicosis, la duración del estado tirotóxico y la condición cardíaca basal (la presencia de cardiopatía isquémica, insuficiencia cardíaca, enfermedad valvular, etc.) más factores de riesgo como la edad avanzada, sexo masculino, hipertensión arterial, diabetes, están relacionados con la inducción de FA74. El desarrollo de la FA en el hipertiroidismo también estaría influenciado por un efecto directo de la hormona, por mayor abundancia de receptores beta en la aurícula, difiriendo la sensibilidad con respecto a los ventrículos y diferencias en la sensibilidad del sistema autonómico entre ambas cámaras75.
En un estudio con pacientes no seleccionados que presentaban FA, menos del 1% de los casos se debía a hipertiroidismo clínico76. Sin embargo, ante la posibilidad de restablecer el eutiroidismo y el ritmo sinusal, está justificada la determinación de TSH en todos los pacientes con FA y otras arritmias supraventriculares sin causa conocida.
El riesgo de tromboembolismo arterial, especialmente el tromboembolismo cerebral, está aumentado en pacientes con hipertiroidismo que padecían FA crónica o paroxística, comparados con pacientes con hipertiroidismo y ritmo sinusal77. Si existe o no un estado de hipercoagulabilidad en el hipertiroidismo aún permanece desconocido. Aunque la concentración en plasma del factor VIII está aumentada, las concentraciones de vitamina K pueden estar disminuidas, y el aumento de la función plaquetaria aún no está demostrada78.
Un importante estudio realizado en hospitales de Dinamarca79 examinó el riesgo de FA en pacientes con hipertiroidismo con edades entre 20 y 89 años, durante un período de 20 años (1977-1999). La muestra tomó 40628 pacientes con diagnóstico de hipertiroidismo, siendo el 84,9% mujeres y aproximadamente 1/3 de los pacientes tenía 70 ó más años. Se diagnosticaron a los 30 días del diagnóstico de hipertiroidismo 3362 casos de FA (8,3%) condición que fue tomada como criterio de inclusión para el estudio. La proporción con FA fue más alta en hombres que en mujeres (12,1% vs 7,6%). Menos del 1% de los pacientes hipertiroideos menores a 40 años tuvo FA, mientras entre el 10% y el 20% de los pacientes mayores de 60 años la presentaron. Entre el 20 a 40% de los pacientes con cardiopatía isquémica, insuficiencia cardíaca congestiva, o enfermedad valvular tuvo FA. El ajuste a odds ratio de la FA fue casi el doble en hombres, y el odds ratio de FA aumentó 1,7 por cada 10 años de edad. Con la presencia de cardiopatía isquémica, insuficiencia cardíaca y enfermedad valvular, el odds ratio para FA aumentó 1,8; 3,9 y 2,6 respectivamente. En la Tabla 4 se resumen los datos más relevantes de este estudio.

El control de la FC es el más importante objetivo de la terapia y puede lograrse con la combinación de fármacos antitiroideos y betabloqueantes80, con lo cual la conducción aurículo-ventricular se retrasa.
Pacientes hipertiroideos con FA muestran a menudo signos de insuficiencia cardíaca derecha (edemas periféricos, congestión hepática) e izquierda (más frecuentemente en personas mayores asociada a alguna enfermedad cardíaca), donde es necesaria la administración de diuréticos. Cuando los niveles de la HT comienzan a disminuir por la influencia del tratamiento antitiroideo, la FA revierte a ritmo sinusal en muchos pacientes.
Algunas experiencias81 indican que en el 62% de los casos de FA, ésta revierte a ritmo sinusal dentro de los primeros 3 ó 4 meses después del control del hipertiroidismo, incluso sin tratamiento antiarrítmico. Y que es poco probable que la FA que persiste más allá de este período revierta a ritmo sinusal sin cardioversión. Fármacos antiarrítmicos como disopiramida (300 mg) usados posteriormente a una cardioversión satisfactoria por un período de 3 meses, evitó la recaída de la FA en los primeros meses, situación que se dio con más frecuencia en aquellos pacientes que no recibieron fármacos antiarrítmicos poscardioversión82.
En pacientes que fueron sometidos a cardioversión, el 89,9% fue restablecido a ritmo sinusal. Con un seguimiento a 5 años el 47,6% se mantenía en ritmo sinusal, tasa elevada comparada a otros estudios de cardioversión que mostraron tasa de recaída entre un 40% y 80% en el primer año83.
Es importante reconocer ante una recaída de FA, la posibilidad de un nuevo episodio de hipertiroidismo, que deberá formar parte del tratamiento la restauración al estado eutiroideo.
Otras arritmias en el hipertiroidismo
Se sabe que las ESV pueden desencadenar una FA, especialmente aquellas que se originan a nivel de las venas pulmonares104, y esto se observa más frecuentemente en los pacientes hipertiroideos que en la población general105. El número de pacientes con taquicardia supraventricular (definida como aquella que tiene mayor a 130 latidos por minuto, y más de 10 ESV en un trazado) disminuye significativamente luego del inicio del tratamiento del hipertiroidismo105, con mayor prevalencia en los individuos mayores de edad. Cuando se realiza un estudio Holter se constata una prevalencia significativa de latidos ectópicos auriculares (ESV mayores a 240 en 24 horas) en comparación a individuos eutiroideos, las cuales persisten por más de tres meses luego de iniciar el tratamiento antitiroideo, y de lograr un estado eutiroideo.
En contraste con las arritmias supraventriculares, las arritmias ventriculares son infrecuentes en la tirotoxicosis y se observan con similar frecuencia que en la población general106. La taquicardia ventricular o la fibrilación ventricular sólo ocurren en los pacientes con tirotoxicosis que padecían una insuficiencia cardíaca o enfermedad cardíaca previa, en su mayoría por enfermedad coronaria107. La diferencia entre las arritmias auriculares y ventriculares es debida a la diferente sensibilidad que presentan ambos tejidos a la HT. Golf y col.108 encontraron que la acción de los betabloqueantes sobre la aurícula derecha es más de dos veces efectiva que el ventrículo izquierdo. También debido a un aumento en la concentración de los canales de potasio dependientes de voltaje (especialmente los Kv 1.5) en la aurícula en comparación al ventrículo109.
Enfermedades cardiovasculares relacionadas con el hipertiroidismo
Insuficiencia cardíaca e hipertiroidismo
Entre las alteraciones cardiovasculares del hipertiroidismo se destacan el aumento del GC en reposo y el aumento de la contractilidad miocárdica. El hipertiroidismo causa comúnmente un estado circulatorio hipercinético como resultado del efecto directo de la HT sobre la contractilidad y la FC, tanto como un efecto indirecto sobre la circulación periférica, conduciendo a un aumento del volumen y vasodilatación. Estos cambios cardiovasculares pueden agravar una enfermedad cardíaca preexistente o directamente conducir a una enfermedad cardíaca tirotóxica.
A pesar de ello, sólo una minoría de los pacientes presenta signos y síntomas indicativos de IC (ocurriendo sólo en un 6% de los pacientes)110, como cardiomegalia, disnea de esfuerzo, ortopnea y disnea paroxística nocturna, o signos como edemas periféricos, distensión de venas cervicales y tercer ruido (R3). Este conjunto de hallazgos, junto con la falta de aumento de la fracción de eyección ventricular con el ejercicio, indican la posibilidad de que exista una miocardiopatía hipertiroidea. El término insuficiencia cardíaca de alto gasto, utilizado a menudo en este contexto, es inadecuado; aunque los síntomas en el ejercicio no parecen deberse a insuficiencia cardíaca, sino más bien a debilidad de la musculatura esquelética2.
En los estados de alto gasto, sin embargo, puede aumentar la reabsorción renal de sodio, con expansión del volumen plasmático y aparición de edemas periféricos, derrame pleural y distensión de las venas del cuello. Llama la atención que, aunque las resistencias disminuyen, el lecho pulmonar no se ve afectado de la misma manera y debido al aumento del GC en la circulación pulmonar, se produce un aumento de las presiones pulmonares. Esto provoca el aumento de la presión venosa media, distensión de venas cervicales, congestión hepática y edemas periféricos asociados a hipertensión pulmonar (HTP) e insuficiencia cardíaca derecha111.
Los pacientes con hipertiroidismo de larga duración y taquicardia o fibrilación auricular importantes, pueden desarrollar bajo GC. La alteración de la contractilidad con fracción de eyección baja, R3, y congestión pulmonar son compatibles con insuficiencia cardíaca congestiva2. Esto se presenta más frecuentemente con enfermedad cardíaca preexistente (cardiopatía isquémica, valvular, hipertensiva, cardiomiopatía alcohólica) y en pacientes mayores112. Se postula que los mecanismos podrían deberse a: 1- que la reducción de la reserva contráctil podría perjudicar la habilidad de aumentar el GC para emparejar el aumento de la demanda metabólica a nivel periférico; 2- una miocardiopatía producida por el exceso de HT, produciendo un miocardio atontado113, llevando a un estrés extremo en el miocito y en algunos casos produciendo necrosis114; 3- una taquicardiomiopatía producida por una prolongada taquicardia sinusal o fibrilación auricular de alta respuesta ventricular. Cuando el ventrículo izquierdo se dilata, puede aparecer también insuficiencia mitral. Es importante reconocer este hecho, ya que los tratamientos que tienen como objetivo reducir la FC o controlar la respuesta ventricular en la FA, parecen mejorar la función ventricular izquierda incluso antes de iniciado el tratamiento antitiroideo2. Dado que estos pacientes se encuentran en situación crítica, deben ser tratados en unidades de cuidados intensivos.
El tratamiento de la IC, muchas veces asociada a fibrilación auricular, incluye reducción de la sobrecarga de volumen con diuréticos endovenosos, como la furosemida, y el control de la FC con digitálicos como la digoxina, que usualmente requerirá una dosis más elevada por situaciones ya comentadas (hipertiroidismo y FA).
Algunos pacientes con hipertiroidismo (como sucede en general, en la insuficiencia cardíaca congestiva) no toleran el inicio del tratamiento con betabloqueantes (BB) en dosis completas, por lo que éste debe iniciarse con dosis bajas de BB de corta duración, los cuales mejoran los síntomas no cardíacos, y además disminuyen la FC, logrando una mejoría de la función ventricular, siempre y cuando la situación clínica lo permita y se piense que la causa predominante de la descompensación cardiológica sea la FC. Los BB están contraindicados en pacientes con marcada hipotensión, bradicardia, o bloqueos aurículoventriculares de segundo o tercer grado. En aquellos que no puedan usar betabloqueantes (enfermedad pulmonar obstructiva crónica -EPOC), los bloqueantes cálcicos no dihidropiridínicos como el diltiazem pueden ser usados, siendo una droga segura y efectiva en mejorar los síntomas hiperadrenérgicos del hipertiroidismo, sin comparación con los BB115.
El tratamiento del síndrome hipertiroideo es inicialmente medicamentoso utilizando betabloqueantes116, tales como propranolol, dado que no sólo disminuye la FC sino también la conversión periférica de T4 a T3. Dentro de las drogas antitiroideas como metilmercaptoimidazol (MMI) o propiltiouracilo117-119, estos dos últimos fármacos comienzan su acción antitiroidea a los 7 días del inicio del tratamiento.
El tratamiento continúa dependiendo de la etiología del síndrome hipertiroideo, si éste está producido por una enfermedad de Graves120,121, se puede continuar con el tratamiento médico o realizar tiroidectomía o iodoradioactividad122-125, salvo que el nódulo sea mayor a 2 cm en cuyo caso será de tratamiento quirúrgico.
En casos severos de hipertiroidismo, se puede utilizar carbonato de litio en dosis de 900 a 1350 mg por 4 a 6 semanas junto a MMI, controlando la función renal y la litemia126-128. También se utiliza el litio si hay contraindicación del MMI.
La utilización de agentes colecistográficos, como el ácido iopanoico, se basa en inhibidores potentes de la 5 desyodasa, la cual cataliza la activación de la prohormona de T4 a T3, reducen la captación periférica de hormonas tiroideas en los tejidos y son inhibidores de la síntesis nuclear de T3. Se utilizan en dosis de 500 mg dos veces al día129.
Hipertensión pulmonar e hipertiroidismo
La hipertensión pulmonar es definida como una presión arterial pulmonar media superior a 25 mm Hg medida en reposo a nivel del mar. La HTP es una entidad rara, progresiva, y a menudo una enfermedad fatal de causa desconocida. Esta enfermedad es más común en mujeres que en hombres (1,7:1) y la edad media de presentación es a los 30 años130. La causa puede ser primaria (idiopática) o secundaria (colagenopatías, virus de inmunodeficiencia humana -VIH-, EPOC, embolismo pulmonar, IC izquierda, abuso de alcohol, apnea del sueño, etc.).
En pocos casos la manifestación prominente cardiovascular en el hipertiroidismo puede presentarse con IC derecha, con grados variables de insuficiencia tricuspídea e hipertensión pulmonar. Posibles mecanismos de estas anormalidades cardíacas incluyen:
- El aumento del volumen sanguíneo o aumento de la carga del ventrículo derecho (VD) por aumento del GC y del retorno venoso.
- Las paredes delgadas del VD, en oposición a las del VI, pueden hacer al VD más susceptible a la sobrecarga de volumen.
- El GC aumentado y un mecanismo autoinmune de la HT producen una injuria a nivel endotelial que puede llevar a hipertensión pulmonar.
- Habría una mayor sensibilidad a las catecolaminas, causando vasoconstricción pulmonar, reducción de la distensibilidad, con aumento en las resistencias.
- El aumento de la resistencia vascular pulmonar podría también resultar de un aumento del metabolismo de ciertas sustancias vasodilatadoras pulmonares (prostaciclinas, óxido nítrico)131, y disminución o alteración en el metabolismo de sustancias vasoconstrictoras (serotonina, endotelina 1 y tromboxano)132.
- Los niveles de la molécula fosfolambano, proteína inhibidora reversible de la actividad ATPasa del retículo sarcoplasmático, estaría disminuida con aumento en la concentración de Ca2+ en el músculo liso, y mayor efecto vasoconstrictor133.
Por otro lado, algunos autores han demostrado que el metimazol parece tener un efecto vasodilatador directo sobre la vasculatura pulmonar al inhibir la producción de una enzima llamada nitro-L-arginina metil ester (L-NAME), induciendo un aumento en la producción de óxido nítrico134. Con respecto a la prevalencia de la HTP asociada al hipertiroidismo, Marvisi y col.135 hallaron una prevalencia del 43% en 114 pacientes hipertiroideos.
Diversos estudios han demostrado que pacientes hipertiroideos con HTP, luego del tratamiento llegando a un estado eutiroideo, logran a los pocos meses una marcada disminución de su HTP (una diferencia de hasta 20 mm Hg) y hasta normalización de las presiones136.
Daño miocárdico activo y hallazgos anatomopatológicos en el hipertiroidismo
La detección no invasiva de daño miocárdico en el hipertiroidismo puede ser posible con anticuerpos monoclonales (indio111 - ibritumimab). La unión de estos anticuerpos a la miosina toma lugar cuando únicamente ocurre disrupción del sarcolema y las células llegan a estar dañadas. Se ha visto que en pacientes con hipertiroidismo, con IC y depresión de la FE, en quienes fue demostrado daño miocárdico con anticuerpos monoclonales antimiosina, luego del tratamiento antitiroideo, tanto el daño como la disfunción miocárdica fueron resueltas (Figura 8). Esto sugiere que el daño miocárdico puede ser un mecanismo concurrente de la IC junto con la sobrecarga de volumen, en pacientes con hipertiroidismo137.
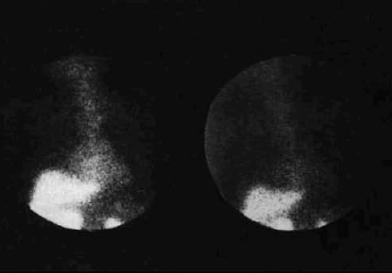
Figura 8. Anticuerpos antimiosina monoclonales marcados con Indio111 en paciente con hipertiroidismo. Izquierda: Al inicio de la presentación clínica, se visualiza en la región en blanco la zona de daño miocárdico. Derecha: meses posteriores al tratamiento se visualiza menor absorción de los anticuerpos indicando recuperación del daño miocárdico115.
En estudios de autopsias de humanos con hipertiroidismo e IC con miocardiopatía dilatada, se ha reportado, en ausencia de otras enfermedades cardíacas, fibrosis intersticial y perivascular, hipertrofia de miocardio, necrosis, y edema celular132 (Figura 9).
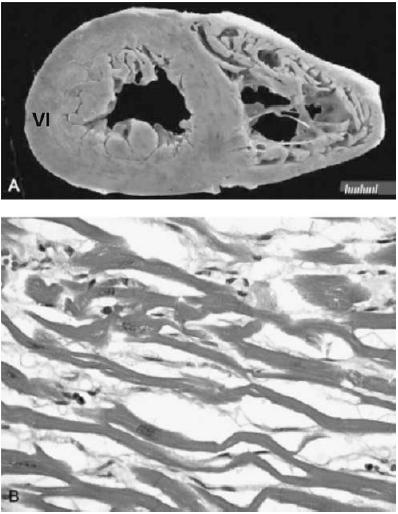
Figura 9. Corazón e hipertiroidismo. A: Corte transversal del corazón que muestra dilatación e hipertrofia biventricular. B: Microfotografía del ventrículo izquierdo (VI) que demuestra edema intersticial y miocitos delgados169.
El hipertiroidismo y el grosor de la pared carotídea
El grosor de la íntima media (IMT) de la carótida, puede ser medido en forma no invasiva a través de la ecografía. El aumento de la IMT se ve asociado con una alta prevalencia de ateroesclerosis y de eventos cardiovasculares finales tales como infarto agudo de miocardio (IAM) o accidente cerebrovascular (ACV)138-140.
Un análisis141 demostró una relación directa entre la función tiroidea y el índice de la IMT. Los participantes que tenían valores de TSH disminuidos y los hipertiroideos, tenían valores de IMT elevados en comparación a los individuos con valores de TSH elevados. Esta relación, se mantuvo estadísticamente significativa luego de realizar los ajustes correspondientes. Los participantes con menor valor de TSH tenían los valores más elevados de IMT, en comparación a los sujetos con nivel de TSH plasmática dentro del segundo y el tercer quartilo.
La asociación entre hipertiroidismo e IMT aumentada puede ser explicada por la vaso dilatación periférica que genera el hipertiroidismo, con la consecuente disminución en la perfusión renal generando la activación del sistema renina-angiotensina-aldosterona142. De esta manera se produce retención de sodio y agua, la angiotensina II por otra parte estimula el crecimiento de células musculares lisas y de la síntesis de colágeno143-145. Por otra parte, se ha demostrado que el tratamiento con T3 genera hipertrofia en las arterias coronarias146. La hipertrofia vascular está asociada a una rigidez vascular incrementada, esta incrementada rigidez se ha visto en la carótida en los pacientes con enfermedad de Graves147-148.
No está claro si hay mecanismos adicionales que contribuyan a aumentar el grosor de la pared arterial en el hipertiroidismo.
El aumento de la IMT puede simplemente reflejar, una respuesta adaptativa de la pared del vaso ante los cambios del shear stress149. Esta situación también podría ser causada en el hipertiroidismo por un aumento de la frecuencia cardíaca y del incremento en la presión de pulso150.
El hipertiroidismo y los efectos en la coagulación y la fibrinólisis
La asociación entre el sistema hemostático y la enfermedad tiroidea es conocida desde hace largo tiempo. La primera asociación clínica fue descripta en el año 1913 cuando Kaliebe describió un episodio de trombosis en la vena cerebral en una paciente con tirotoxicosis151. Varios elementos se ven involucrados en la formación de trombos152. Tanto la disfunción tiroidea como las enfermedades autoinmunes pueden modificar el proceso primario o secundario de la hemostasis llevando a sangrado o por el contrario trombosis. La púrpura trombocitopénica idiopática, el síndrome antifosfolipídico o la hemofilia adquirida se han asociado en casos de enfermedad tiroidea autoinmune153.
La influencia de las hormonas tiroideas en el sistema de la coagulación y de la fibrinólisis está mediada principalmente por la interacción que existe entre estas hormonas y sus receptores154. Diversas anormalidades han sido descriptas desde anormalidades subclínicas solamente evidenciadas por el laboratorio hasta eventos tromboembólicos o hemorrágicos fatales155.
En el hipertiroidismo se observa un incremento en el riesgo de trombosis en las venas cerebrales y del seno151.
Tanto en el hipotiroidismo como en el hipertiroidismo se observa un disbalance entre la coagulación y el sistema fibrinolítico, ambos grupos de enfermos presentan test de laboratorio modificados en la coagulación y en la fibrinólisis. No hay datos disponibles hasta el momento del grado de hipercoagulabilidad que se evidencia en el hipertiroidismo. El complejo del balance hemostático tal vez estaría mediado por factores autoinmunes. Otra hipótesis podría implicar anormalidades en el sistema adrenérgico o el sistema de vasopresina-arginina156,157.
Algunos test de laboratorio han descripto alteraciones en el hipertiroidismo subclínico, sólo un estudio de baja calidad, ha investigado extensamente las anormalidades de la coagulación- fibrinólisis en el hipertiroidismo subclínico158. Muchos médicos aún ignoran la relación que existe entre las hormonas tiroideas y el sistema de la coagulación, y es fundamental comprender que el balance hemostático se ve afectado en la disfunción tiroidea.
Hipertiroidismo subclínico: su afectación en la calidad de vida y la morfología cardíaca en jóvenes y edad media
El hipertiroidismo subclínico se caracteriza por la presencia de baja o indetectable TSH y valores normales de HT periféricas. El impacto del mismo aún no ha sido establecido, los pacientes con hipertiroidismo subclínico muestran pocos y no específicos síntomas y signos de hipertiroidismo.
El hipertiroidismo subclínico puede ser de causa endógena o exógena. Este último se presenta frecuentemente en pacientes tratados con dosis excesivas de T4 para sus requerimientos, ya sea por una enfermedad nodular tiroidea o postiroidectomía, y en pacientes tratados por un cáncer de tiroides con dosis inhibitorias de HT. También puede verse en pacientes con un bocio multinodular o un nódulo tiroideo funcionante.
Se ha visto que el hipertiroidismo afecta la función cardíaca y su morfología84,85. Se observa en estos pacientes un aumento de la FC, un aumento de la función sistólica del VI y una alteración en la función diastólica. También se observa en estos pacientes un incremento de la masa del VI86-94, como consecuencia del aumento crónico en la poscarga y es responsable de la disfunción diastólica del VI y de la falta de respuesta ante el ejercicio92,93. Estas anormalidades se ven revertidas ante el uso de betabloqueantes95. La HVI es un factor independiente de morbimortalidad cardiovascular96. Cuando se realiza un Holter, en los hipotiroideos subclínicos, se observa un aumento en la prevalencia de extrasístoles supraventriculares (ESV), episodios de taquicardia por reentrada nodal89 y taquicardia sinusal. En estos pacientes se observa un estímulo directo en el nódulo sinusal y atrial97. La regulación de FC por las HT es compleja: en parte un efecto indirecto sobre el sistema adrenérgico y por otra parte una acción directa sobre cronotropismo98,99. En ratas se observa la acción sobre los niveles del ARNm de los canales relacionados al potasio, y recientemente se ha visto que los canales HCN2 son dependientes de las HT100.
El significado de una FC elevada en individuos jóvenes o de edad media es discutido, por el contrario en los de edad avanzada está asociado a un incremento en mortalidad cardiovascular y no cardiovascular101. El tratamiento estaría indicado en este último grupo de pacientes aún con síntomas leves de tirotoxicosis y factores de riesgo cardiovascular o un bocio de gran tamaño, debido a que puede terminar desarrollando FA, finalmente incrementando el riesgo de eventos tromboembólicos.
Por otra parte, en los jóvenes o en los de edad media el tratamiento debería implementarse para mejorar la calidad de vida y evitar las consecuencias a largo plazo ante la exposición de elevadas concentraciones de HT.
La prevalencia del hipertiroidismo subclínico en la comunidad se observa entre el 0,5 y el 0,8% en la población yodo suficiente; en sujetos mayores de 60 años es del 3 al 6%.
El hipertiroidismo subclínico es más común que el hipertiroidismo en personas mayores de 60 años y en la mayoría de los casos no progresa a hipertiroidismo102, siendo del 1% por año, y en los de menor edad del 1 al 5% por año.
Johann y colaboradores103 estudiaron a 23638 personas para saber si el hipertiroidismo subclínico era un factor de riesgo para FA, clasificándolas de acuerdo a las concentraciones de TSH, T3 y T4. El grupo 1 (n=22300) estuvo compuesto por personas con valores normales tanto de TSH, T3 y T4. El grupo 2 (n=725) fueron pacientes con hipertiroidismo (baja TSH, alta T3 y T4). El grupo 3 (n=613) eran pacientes con hipertiroidismo subclínico (baja TSH, con T3 y T4 normales). Los resultados fueron los siguientes: la FA estuvo presente en 513 personas (2,3%) en el grupo 1, en 100 personas (13,8%) en el grupo 2 y en 78 personas (12,7%) en el grupo 3. El riesgo relativo de FA en sujetos con hipertiroidismo subclínico comparado con sujetos con estado eutiroideo fue de 5,2 (IC 95% 2,2-8,7, p<0,01).
Discusión
Desde hace más de 200 años, se reconoce la relación existente entre la HT, el corazón y el sistema cardiovascular periférico. La HT tiene relevantes acciones sobre el corazón y la circulación, donde genera múltiples cambios incluyendo alteraciones hemodinámicas y efectos mediados sobre el miocito cardíaco a través de la expresión genómica y no genómica.
Ejemplo de estos efectos son mencionados por los investigadores Merillon y Feldman donde cada uno, con respecto al aumento de la contractilidad miocárdica, defiende diferentes posibles mecanismos (el primero a favor del aumento de la FC y la precarga con el consecuente aumento del GC, el segundo mencionando el efecto directo de la HT sobre el aumento de la velocidad en el acortamiento del músculo cardíaco independientemente de las condiciones de carga), los cuales no se contraponen, sino más bien se complementan como consecuencia de la acción de la HT a nivel periférico y miocárdico.
Con respecto al resto de los mecanismos fisiopatológicos, no se han encontrado grandes controversias, muchos de los cuales tienen su demostración en el campo de la investigación científica y si bien otras explicaciones de estos son hipótesis, los razonamientos son coherentes; aún hay mecanismos desconocidos y seguramente en línea de investigación.
La cardioversión debe realizarse 4 meses posteriores al estado eutiroideo (ya sea con medicación o en el mejor de los casos con radioablación de la glándula con yodo131), sabiendo que más del 50% de los casos revierte espontáneamente a ritmo sinusal. En un estudio retrospectivo de pacientes con hipertiroidismo, se observó que el principal factor de embolias era la edad, más que la presencia de FA. El análisis retrospectivo de series extensas de pacientes no halló una prevalencia de episodios tromboembólicos superior al riesgo de hemorragias graves descripto con el tratamiento con warfarina159.
En la literatura, se publican estudios donde el tratamiento en pacientes con insuficiencia cardíaca gatillada por el hipertiroidismo muestra una potencial mejoría y reversión del cuadro postratamiento: Umpierrez y col.160 publicaron un estudio de 7 pacientes con hipertiroidismo e IC en quienes la media de la FE aumentó de 28 a 55% después del tratamiento antitiroideo. La FE se normalizó en 5 pacientes, y una mejoría de severa a leve en los 2 pacientes restantes. Goldman y col.161 presentaron el caso de un paciente de 56 años con IC que tenía una hipocinesia global y una FE del 35% por ecocardiograma, que aumentó a un 55% en 3 semanas después del tratamiento antitiroideo. Kantharia y col.162 reportaron una mujer de 52 años con IC con una hipocinesia global y una FE del 30% en la admisión con una notable mejoría con el tratamiento antitiroideo (FE=58%).
Con respecto a la hipertensión pulmonar ocasionada por estados de hipertiroidismo, Merce y col. publicaron un estudio ecocardiográfico prospectivo de 39 pacientes con hipertiroidismo comparados con un grupo control, revelando que la presión media de la arteria pulmonar fue significativamente mayor en los pacientes hipertiroideos (38 vs 27 mm Hg) además de presentar más casos de insuficiencia tricuspídea moderada a severa que en el grupo control (7 vs 1). Este estudio y otros sugieren que los casos de insuficiencia cardíaca derecha asociada a hipertensión pulmonar son muy comunes y muchas veces subdiagnosticados.
Por ello, se debe realizar un ecocardiograma de rutina a los pacientes con hipertiroidismo para pueden ser identificar precozmente a estas anormalidades163.
Todos los estudios coinciden que la terapéutica final y óptima en el hipertiroidismo con afectación cardiovascular es la ablación de la glándula con yodo131, donde en la mayoría de los casos los trastornos anatómicos y hemodinámicos revierten hacia la normalidad, al lograr estados de eutiroidismo.
Conclusión
La enfermedad tiroidea es bastante común, con una prevalencia del 9% al 15% en la población adulta164. Los signos y síntomas cardiovasculares son algunos de los más importantes y relevantes que acompañan al hipertiroidismo. El entendimiento de los mecanismos celulares de la acción de la HT sobre el corazón y el sistema cardiovascular, explican los cambios que se producen en el gasto cardíaco, la contractilidad, la presión arterial, la resistencia vascular, y trastornos en el ritmo y que resultan de la disfunción de la glándula. La importancia del reconocimiento de los efectos del hipertiroidismo sobre el corazón, también deriva de la observación que la restauración a la normalidad de la función tiroidea muy a menudo revierte los trastornos hemodinámicos cardiovasculares. Es el objetivo principal de esta revisión esclarecer el tema para un apropiado diagnóstico y tratamiento que debe ejercer el profesional clínico ante su aparición, ya que estos trastornos pueden agravar una enfermedad cardíaca establecida o conducir a una afectación cardíaca por sí misma165-167.
Este tema por cierto, investigado desde hace mucho tiempo, no deja de ser fascinante, con mecanismos fisiopatológicos aún desconocidos en contraste con la terapéutica eficaz, en su repercusión clínica. Estos pacientes deben ser tratados en forma multidisciplinaria, es muy importante que se unan para el tratamiento los cardiólogos y los endocrinólogos.
1. Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl Med 2001;344:501-508. [ Links ]
2. Dillmann W. Biochimical basis of thyroid hormone action in the heart. Am J Med 1990;88:626-630. [ Links ]
3. Perel C, Echin M. Insuficiencia cardíaca y tiroides. Daño miocárdico en el hipertiroidismo. Insuf Card 2006;1(1):43-51. [ Links ]
4. Harvey CB, Williams GR. Mechanism of thyroid hormone action. Thyroid 2002;12:441-446. [ Links ]
5. Dillmann WH. Cellular action of thyroid hormone on the heart. Thyroid 2002;12:447-452. [ Links ]
6. Danzi S, Klein I. Thyroid hormone-regulated cardiac gene expression and cardiovascular disease. Thyroid 2002;12:467-472. [ Links ]
7. Danzi S, Ojamaa K, Klein I. Triiodothyronine-mediated myosin heavy chain gene transcription in the heart. Am J Physiol Heart Circ Physiol 2003;284:2255-2262. [ Links ]
8. Morkin E. Control of cardiac myosin heavy chain gene expression. Micros Res Tech 2000;50:522-531. [ Links ]
9. Reiser PJ, Portman MA, Ning X, et al. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am J Physiol 2001;280:1814-1820. [ Links ]
10. Ojamaa K, Ascheim D, Hryniewics K, et al. Thyroid hormone therapy of cardiovascular disease. Cardiovasc Rev Rep 2002;23:20-26. [ Links ]
11. Carr A, Kranias E. Thyroid hormone regulation of calcium cycling proteins. Thyroid 2002;12:453-457. [ Links ]
12. Ojamaa K, Kenessey A, Klein I. Thyroid hormone regulation of phospholamban phosphorylation in the rat heart. Endocrinology 2000;141:2139-2144. [ Links ]
13. Biondi B, Palmieri E, Lombardi G, et al. Subclinical hypothyroidism and cardiac function. Thyroid 2002;12:505-510. [ Links ]
14. Virtanen V, Saha H, Groundstroem K, et al. Thyroid hormone substitution therapy rapidly enhances left-ventricular diastolic function in hypothyroid patients. Cardiology 2001;96:59-64. [ Links ]
15. Davis P, Davis F. Nongenomic actions of thyroid hormone on the heart. Thyroid 2002;12:459-466. [ Links ]
16. Demers L, Spencer C. Laboratory medicine practice guidelines, laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid 2003;13:3-126. [ Links ]
17. Cooper D, et al. Management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Guidelines task force thyroid 2006;16:21-34. [ Links ]
18. Hoit B, Khoury S, Shao Y, et al. Effects of thyroid hormone on cardiac beta-adrenergic responsiveness in conscious baboons. Circulation 1997;96:592-598. [ Links ]
19. Ojaama K, Klein I, Sabet A, et al. Changes in adenylyl cyclase isoforms as a mechanism for thyroid hormone modulation of cardiac beta-adrenergic receptor responsiveness. Metabolism 2000;49:275-279. [ Links ]
20. Ventrella S, Klein I. Beta-adrenergic receptor blocking drugs in the management of hyperthyroidism. The Endocrinologist 1994;4:391-399. [ Links ]
21. Hartong R, Wang N, Kurokawa R, Lazar MA, Glass CK, Apriletti, Dillmann WH. Delineation of three different thyroid hormoneresponse elements in promoter of rat sarcoplasmic reticulum Ca2-ATPase gene. J Biol Chem 1994;269:13021-13029. [ Links ]
22. Braunwald E, Sonnenblick EH, Ross Jr J. Mechanisms of cardiac contraction and relaxation. In: Braunwald E, ed. Heart disease: a textbook of cardiovascular medicine, 4th edition. Philadelphia: W.B. Saunders Company; 1992;351-392. [ Links ]
23. Kahaly GJ, Wagner S, Nieswandt J, Mohr-Kahaly S, Ryan TJ. Stress echocardiography in hyperthyroidism. J Clin Endocrinol Metab 1999;84:2308-2313. [ Links ]
24. Merillon JP, Passa P, Chastre J, Wolf A, Gourgon R. Left ventricular function and hyperthyroidism. Br Heart J 1981;46:137-143. [ Links ]
25. Feldman T, Borow KM, Sarne DH, Neumann A, Lang RM. Myocardial mechanics in hyperthyroidism: importance of left ventricular loading conditions, heart rate and contractile state. J Am Coll Cardiol 1986;7:967-974. [ Links ]
26. McDevitt D, Riddell J, Hadden D, Montgomery D. Catecholamine sensitivity in hyperthyroidism and hypothyroidism. Br J Clin Pharmacol 1978;6:297-301. [ Links ]
27. Kosar F, Sahin I, Turan N, Topal E, Aksoy Y, Taskapan C. Evaluation of right and left ventricular function using pulsed-wave tissue Doppler echocardiography in patients with subclinical hypothyroidism. J Endocrinol Invest 2005;28:704-710. [ Links ]
28. Franzoni F, Galetta F, Fallahi P, Tocchini L, Merico G, Braccini L, Rossi M, Carpi A, Antonelli A, Santoro G. Effect of L-thyroxine treatment on left ventricular function in subclinical hypothyroidism. Biomed Pharmacother 2006;60:431-436. [ Links ]
29. Arinc H, Gunduz H, Tamer A, Seyfeli E, Kanat M, Ozhan H, Akdemir R, Uyan C. Tissue Doppler echocardiography in evaluation of cardiac effects of subclinical hypothyroidism. Int J Cardiovasc Imaging 2006;22:177-186. [ Links ]
30. Vitale G, Galderisi M, Lupoli GA, Celentano A, Pietropaolo I, Parenti N, de Divitiis O, Lupoli G. Left ventricular myocardial impairment in subclinical hypothyroidism assessed by a new ultrasound tool: pulsed tissue Doppler. J Clin Endocrinol Metab 2002;87:4350-4355. [ Links ]
31. Monzani F, Di Bello V, Caraccio N, Bertini A, Giorgi D, Giusti C, Ferrannini E. Effect of levothyroxine on cardiac function and structure in subclinical hypothyroidism: a double blind, placebocontrolled study. J Clin Endocrinol Metab 2001;86:1110-1115. [ Links ]
32. Aghini-Lombardi F, Di Bello V, Talini E, Di Cori A, Monzani F, Antonangeli L. Early textural and functional alterations of left ventricular myocardium in mild hypothyroidism. Eur J Endocrinol 2006;155:3-9. [ Links ]
33. Fazio S, Palmieri EA, Lombardi G, Biondi B. Effects of thyroid hormone on the cardiovascular system. Recent Prog Horm Res 2004;59:31-50. [ Links ]
34. Osman F, Gammage MD, Franklyn JA. Thyroid disease and its treatment: short-term and long-term cardiovascular consequences. Curr Opin Pharmacol 2001;1:626-631. [ Links ]
35. Biondi B, Palmieri EA, Fazio S, Cosco C, Nocera M, Sacca L, Filetti S, Lombardi G, Perticone F. Endogenous subclinical hyperthyroidism affects quality of life and cardiac morphology and function in young and middle-aged patients. J Clin Endocrinol Metab 2000;85:4701-4705. [ Links ]
36. Smit JW, Eustatia-Rutten CF, Corssmit EP, Pereira AM, Frolich M, Bleeker GB, Holman ER, van der Wall EE, Romijn JA, Bax JJ. Reversible diastolic dysfunction after long-term exogenous subclinical hyperthyroidism: a randomized, placebo-controlled study. J Clin Endocrinol Metab 2005;90:6041-6047. [ Links ]
37. Hammas RL, Khan NU, Nekkanti R, Movahed A. Diastolic heart failure and left ventricular diastolic dysfunction: what we know and what we don't know! Int J Cardiol 2007;115:284-292. [ Links ]
38. Dillmann W. Diabetes and thyroid hormone-induced changes in cardiac function and their molecular basis. Annu Rev Med 1989;40:373-394. [ Links ]
39. Mintz G, Pizzarello R, Klein I. Enhanced left ventricular diastolic function in hyperthyroidism: Non invasive assessment and response to treatment. J Clin Endocrinol Metab 1991;73:146-150. [ Links ]
40. Rohrer D, Dillman W. Thyroid hormone markedly increases the mRNA coding for sarcoplasmic reticulum Ca2+ ATPase in the rat heart. J Biol Chem 1988;263:6941-6944. [ Links ]
41. Schaefer S, Taylor AL, Lee HR, Niggemann EH, Levine BD, Popma JJ, Mitchell JH, Hillis LD. Effect of increasing heart rate on left ventricular performance in patients with normal cardiac function. Am J Cardiol 1988;61:617-620. [ Links ]
42. Valente M, De Santo C, de Martino Rosaroll D, Di Maio V, Di Meo, De Leo T. The direct effect of the thyroid hormone on cardiac chronotropism. Arch Int Physiol Biochim 1988;97:400-431. [ Links ]
43. Arnosdorf M, Childers R. Atrial electrophysiology in experimental hyperthyroidism in rabbits. Circ Res 1970;26:575-581. [ Links ]
44. Branwald E. Tratado de Cardiología: Texto de Medicina. 7ª ed. Madrid. Elsevier; 2006:2056-2060. [ Links ]
45. Gibson J, Harris A. Clinical studies on the blood volume: V. Hyperthyroidism and myxedema. J Clin Invest 1939;18:59-65. [ Links ]
46. Anthonisen P, Holst E, Thomsen A. Determination of cardiac output and other hemodynamic data in patients with hyper- and hypothyroidism, using dye dilution technique. Scand J Clin Lab Invest 1960;12:472-480. [ Links ]
47. Resnick L, Laragh J. Plasma renin activity in syndromes of thyroid hormone excess and deficiency. Life Sci 1982;30:585-586. [ Links ]
48. Ellis LB, Mebane JG, Maresh G, Hultgren HN, Bloomfield RA. The effect of myxedema on the cardiovascular system. Am Heart J 1952;43(3):341-356. [ Links ]
49. Biondi B, Palmieri E, Lombardi G, et al. Effects of thyroid hormone on cardiac function: The relative importance of heart rate, loading conditions, and myocardial contractility in the regulation of cardiac performance in human hyperthyroidism. J Clin Endocrinol Metab 2002;87:968-974. [ Links ]
50. Rothfeld J, LeWinter M, Tishler M. Left ventricular systolic torsion and early diastolic filling by echocardiography in normal humans. Am J Cardiol 1998;81:1465-1469. [ Links ]
51. Toft A, Boon N. Thyroid disease and the heart. Heart 2000;84:455-460. [ Links ]
52. Ojamaa K, Klemperer JD, Klein I. Acute effects of thyroid hormone on vascular smooth muscle. Thyroid 1996;6:505-512. [ Links ]
53. Streeten D, Anderson Jr G, Howland T, Chiang R, Smulyan H. Effects of thyroid function on blood pressure. Recognition of hypothyroid hypertension. Hypertension 1988;11:78-83. [ Links ]
54. Wilkinson I, MacCallum H, Flint L, Cockcroft J, Newby D, Webb D. The influence of heart rate on augmentation index and central arterial pressure in humans. J Physiol 2000;525:263-270. [ Links ]
55. Bengel F, Lehnert J, Ibrahim T, et al. Cardiac oxidative metabolism, function, and metabolic performance in mild hyperthyroidism: a non-invasive study using positron emission tomography and magnetic resonance imaging. Thyroid 2003;13:471-477. [ Links ]
56. Klemperer J, Klein I, Gomez M, et al. Thyroid hormone treatment after coronary-artery bypass surgery. N Eng J Med 1995;333:1522-1527. [ Links ]
57. Theilen E, Wilson W. Hemodynamic effects of peripheral vasoconstriction in normal and thyrotoxic subjects. J Appl Physiol 1967;22:207-210. [ Links ]
58. Danzi S, Klein I. Thyroid hormone and blood pressure regulation. Curr Hypertens Rep 2003;5:513-520. [ Links ]
59. Weetman A. Graves' disease. N Engl J Med 2000;343:1236-1248. [ Links ]
60. Cacciatori V, Bellavere F, Pessarosa A, et al. Power spectral analysis of heart rate in hyperthyroidism. J Clin Endocrinol Metab 1996;81:2828-2835. [ Links ]
61. Kahaly G, Kampmann C, Mohr-Kahaly S. Cardiovascular hemodynamics and exercise tolerance in thyroid disease. Thyroid 2002;12:473-478. [ Links ]
62. Kahaly G, Dillman W. Thyroid hormone action in the heart. Endocrine Rev 2005;26:704-728. [ Links ]
63. Danzi S, Klein I. Thyroid hormone and the cardiovascular system. Minerva Endocrinologia 2004;29:139-150. [ Links ]
64. Osman F, Franklyn J, Holder R, Sheppard M, Gammage M. Cardiovascular symptoms and cardiac rate and rhythm abnormalities improve with treatment in patients with hyperthyroidism. J Am Coll Cardiol 2007;49:71-81. [ Links ]
65. Lerman J, Means HJ. Cardiovascular symptomatology in exophtalmic goiter. Am Heart J 1932;8:55-65. [ Links ]
66. Valcavi R, Menozzi C, Roti E, et al. Sinus node function in hyperthyroid patients. J Clin Endocrinol Metab 1992;75:239-242. [ Links ]
67. Biondi B, Cooper DS. The clinical significance of subclinical thyroid dysfunction. Endocr Rev 2008;29(1):76-131. [ Links ]
68. Nakazawa H, Lythall D, Noh J, Ishikawa N, et al. Is there a place for the late cardioversion of atrial fibrillation? Along-term follow-up study of patients with post-thyrotoxic atrial fibrillation. Eur Heart J 2000;21:327-333. [ Links ]
69. Frost L, Engholm G, Johnsen S, Moller H, et al. Incident thromboembolism in the aorta and the renal, mesenteric, pelvic, and extremity arteries after discharge from the hospital with a diagnosis of atrial fibrillation. Arch Intern Med 2001;161:272-276. [ Links ]
70. Frost L, Engholm G, Johnsen S, Moller H, et al. Incident stroke after discharge from the hospital with a diagnosis of atrial fibrillation. Am J Med 2000;108:36-40. [ Links ]
71. Auer J, Scheibner P, Mische T, et al. Subclinical hyperthyroidism as a risk factor for atrial fibrillation. Am Heart J 2001;142:838-842. [ Links ]
72. Aras D, Maden O, Ozdemir O, Aras S, Topaloglu S, Yetkin E, Demir AD, Soylu MO, Erdogan MF, Kisacik HL, Korkmaz S. Simple electrocardiographic markers for the prediction of paroxysmal atrial fibrillation in hyperthyroidism. Int J Cardiol 2005;99(1):59-64. [ Links ]
73. Taeko S, Saori K, Jaduk Y, et al. Hyperthyroidism and the management of atrial fibrillation. Thyroid 2002;12:489-493. [ Links ]
74. Martin WH III, Spina RJ, Korte E. Effect of hyperthyroidism of short duration on cardiac sensitivity to beta-adrenergic stimulation. J Am Coll Cardiol 1992;19:185-191. [ Links ]
75. Forfar J. Atrial fibrillation and the pituitary-thyroid axis: a reevaluation. Heart 1997;77:3-4. [ Links ]
76. Presti C, Hart R. Thyrotoxicosis, atrial fibrillation, and embolism, revisited. Am Heart J 1989;273:1057-1061. [ Links ]
77. Loeliger E, Mattern M, Hemker H, et al. The biological disappearance rate of prothrombin, factors VII, IX and X from plasma in hypothyroidism, hyperthyroidism, and during fever. Thromb Diath Haemorrh 1964;10:267-277. [ Links ]
78. Frost L, Vestergaard P, Mosekilde L. Hyperthyroidism and risk of atrial fibrillation or flutter. Arch Intern Med 2004;164:1675-1678. [ Links ]
79. Falk R. Atrial fibrillation. N Engl J Med 2001;344:1067-1078. [ Links ]
80. Kim D, Smith T. Effects of thyroid hormone on sodium pump sites, sodium content and contractile responses to cardiac glycosides in cultured chick ventricular cells. J Clin Invest 1984;74:1481-1488. [ Links ]
81. Nakazawa K, Sakurai K, Hamada N, Momotani N, Ito K. Management of atrial fibrillation in the post-thyrotoxic state. Am J Med 1982;72:903-906. [ Links ]
82. Nakazawa H, Ishikawa N, Noh J, Sugimoto T, Yoshimoto M, Yashiro T, Ozaki O, Ito K. Efficacy of disopyramide in conversion and prophylaxis of post-thyrotoxic atrial fibrillation. Eur J Clin Pharmacol 1991;40:215-219. [ Links ]
83. Klein AL, Grimm RA, Murray RD, Apperson-Hansen C, Asinger RW, Black IW, et al. Use of transesophageal echocardiography to guide cardioversion in patients with atrial fibrillation. N Engl J Med 2001;344:1411-1420. [ Links ]
84. Klein I. Thyroid hormone and the cardiovascular system. Am J Med 1990;88:631-637. [ Links ]
85. Polikar R, Burger AG, Scherrer U, Nicod P. The thyroid and heart. Circulation 1993;87:1453-1441. [ Links ]
86. Bell GM, Sawers SA, Forfar JC, Doig A, Toft D. The effect of minor increments in plasma thyroxine on heart rate and urinary sodium excretion. Clin Endocrinol 1983;18:511-516. [ Links ]
87. Biondi B, Fazio S, Carella C, et al. Cardiac effects of long-term thyrotropin-suppressive therapy with levothyroxine. J Clin Endocrinol Metab 1993;77:334-338. [ Links ]
88. Polikar R, Feld GK, Dittrick FH, Smith J, Nicod P. Effect of thyroid replacement therapy on the frequency of benign atrial and ventricular arrhythmias. J Am Coll Card 1989;14:999-1002. [ Links ]
89. Biondi B, Fazio S, Coltorti F, et al. Reentrant atrioventricular nodal tachycardia induced by levothyroxine. J Clin Endocrinol Metab 1998;83:2643-2645. [ Links ]
90. Ching GW, Franklyn JA, Stallard TJ, Daykin J, Sheppard M, Gammage MD. Cardiac hypertrophy as a result of long-term thyroxine therapy and thyrotoxicosis. Heart 1996;75:363-368. [ Links ]
91. Shapiro LE, Sievert R, Ong L, et al. Minimal cardiac effects in asymptomatic athyreotic patients chronically treated with thyrotropin-suppressive doses ofL-thyroxine. J Clin Endocrinol Metab 1997;82:2592-2595. [ Links ]
92. Fazio S, Biondi B, Carella C, et al. Diastolic dysfunction in patients on thyroid-stimulating-hormone suppressive therapy with levothyroxine: beneficial effect of beta blockade. J Clin Endocrinol Metab 1995;80:2222-2226. [ Links ]
93. Biondi B, Fazio S, Cuocolo A, et al. Impaired cardiac reserve and exercise capacity in patients receiving long-term thyrotropin suppressive therapy with levothyroxine. J Clin Endocrinol Metab 1996;81:4224-4228. [ Links ]
94. Mercuro G, Panzuto MG, Bina A, et al. Cardiac function, physical exercise capacity, and quality of life during long-term thyrotropinsuppressive therapy with levothyroxine: effect of individual dose tailoring. J Clin Endocrinol Metab 2000;85:159-164. [ Links ]
95. Biondi B, Fazio S, Carella C, et al. Control of adrenergic over activity by beta-blockade improves the quality of life in patients receiving long-term suppressive therapy with levothyroxine. J Clin Endocrinol Metab 1994;78:1028-1033. [ Links ]
96. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli W. Prognostic implication of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990;322:1561-1566. [ Links ]
97. Boutin JM, Matte R, D'Amour P, et al. Characteristic of patients with normal T3 and T4 and low TSH response to TRH. Clin Endocrinol 1986;25:579-588. [ Links ]
98. Abe A, Yamamoto T, Isome M, et al. Thyroid hormone regulates expression of Shaker-related potassium channel mRNA in rat heart. Biochem Biophys Res Commun 1998;245:226-230. [ Links ]
99. Williams LT, Lefkowitz RJ, Watanabe AM, Hathaway DR, Besch Jr HR. Thyroid hormone regulation of beta-adrenergic number. J Biol Chem 1977;252:2787-2789. [ Links ]
100. Pachucki J, Burmeister L, Larsen PR. Thyroid hormone regulated hyperpolarization-activated cyclic nucleotide-gated channel (HCN2) mRNA in the rat heart. Circ Res 1999;85:498-503. [ Links ]
101. Kannel WB, Kannel C, Paffenbarger Jr RS, Cupples LA. Heart rate and cardiovascular mortality: The Framingham Study. Am Heart J 1987;113:1489-1494. [ Links ]
102. Utiger R. Subclinical hyperthyroidism -just a low serum thyrotropin concentration, or something more? N Engl J Med 1994;77:332-333. [ Links ]
103. Benjamín E, Levy D, Vaziri S, et al. Independent risk factors for atrial fibrillation in a population-based cohort: the Framingham Heart Study. JAMA 1994;2(71):840-844. [ Links ]
104. Auer J, Berent R, Weber T, Eber B. Hyperthyroidism. Lancet 2003;362:1584. [ Links ]
105. Haissaguerre M, Jais P, Shah D, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 1998;339:659-666. [ Links ]
106. Olshausen K, Bischoff S, Kahaly G, Mohr-Kahaly S, Erbel R, Beyer J, Meyer J. Cardiac arrhythmias and heart rate in hyperthyroidism. Am J Cardiol 1989;63:930-933. [ Links ]
107. Osman F, Daykin J, Sheppard M, Franklyn J, Gammage M. Cardiac rhythm abnormalities in thyrotoxicosis. The explanation for excess vascular mortality. J Endocrinol 2000;164:321. [ Links ]
108. Polikar R, Burger AG, Scherrer U, Nicod P. The thyroid and the heart. Review. Circulation 1993;87:1435-1441. [ Links ]
109. Golf S, Lovstad R, Hansson V. Beta-adrenoceptor density and relative number of beta-adrenoceptor subtypes in biopsies from human right atrial, left ventricular and right ventricular myocardium. Cardiovasc Res 1985;19:636-641. [ Links ]
110. Ojamaa K, Sabet A, Kenessey A, Shenoy R, Klein I. Regulation of rat cardiac Kv1.5 gene expression by thyroid hormone is rapid and chamber specific. Endocrinology 1999;140:3170-3176. [ Links ]
111. Summers V, Surtees S. Thyrotoxicosis and heart disease. Acta Med Scand 1969;169:661-671. [ Links ]
112. Duan Y, Peng W, Wang X, Tang W, Liu X, Xu S, Mao X, Feng S, Feng Y, Qin Y, et al. Community-based study of the association of subclinical thyroid dysfunction with blood pressure. Endocr 2009;35(2):136-142. [ Links ]
113. Nordyke R, Gilbert F, Harada A. Graves' disease: influence of age on clinical findings. Arch Intern Med 1988;148:626-631. [ Links ]
114. Pereira N, Parisi A, Dec G, et al. Myocardial stunning in hyperthyroidism. Clin Cardiol 2000;23:298-300. [ Links ]
115. Marti V, Ballester M, Obrador D, et al. Active myocardial damage in hyperthyroidism. A concurrent mechanism of heart failure reversed by treatment. Eur Heart J 1995;16:1014-1016. [ Links ]
116. Roti E, Montermini M, Roti S, et al. The effect of diltiazem, a calcium channel blocking drug, on cardiac rate and rhythm in hyperthyroid patients. Arch Intern Med 1988;148:919-921. [ Links ]
117. Franklyn J, Maisonneuve P, Sheppard M, Betteridge J, Boyle P. Mortality after the treatment of hyperthyroidism with radioactive iodine. N Engl J Med 1998;38:712-718. [ Links ]
118. Woeber K. Update on the management of hyperthyroidism and hypothyroidism. Arch Intern Med 2000;160:1067-1070. [ Links ]
119. Hasnain M, Khandwala, et al. A case of congestive heart failure due to reversible dilated cardiomyopathy caused by hyperthyroidism. Southern Med J 2004;97;10:1001-1003. [ Links ]
120. Eiffel JA, Estes III NA, Waldo AL, Prystowsky EN, DiBianco R. A consensus report on antiarrhythmic drug use. Clin Cardiol 1994;17:103-116. [ Links ]
121. Newnham HH, Topliss DJ, Le Grand BA, Chosich N, Harper RW, Stockigt JR. Amiodarone-induced hyperthyroidism: assessment of the predictive value of biochemical testing and response to combined therapy using propylthiouracil and potassium perchlorate. Aust NZ J Med 1988;18:37-44. [ Links ]
122. Donaghue KC, Clarke P, Hooper MJ. Amiodarone. The dilemma of hyperthyroxinemia and the treatment of thyrotoxicosis. Med J Aust 1985;142:594-596. [ Links ]
123. Wu SY, Shyh TP, Chopra IJ, Huang HW, Chy PC. Comparison of sodium ipodate (Oragrafin) and propylthiouracil in early treatment of hyperthyroidism. J Clin Endocrinol Metab 1982;54:630-634. [ Links ]
124. Shen DC, Wu SY, Chopra IJ, et al. Long term treatment of Graves' hyperthyroidism with sodium ipodate. J Clin Endocrinol Metab 1985;61:723-727. [ Links ]
125. Hopra IJ, Van Herle AJ, Korenman SG, Viosca S, Younai S. Use of sodium ipodate in management of hyperthyroidism in subacute thyroiditis. J ClinEndocrinol Metab 1995;80:2178-2180. [ Links ]
126. Awin CT, Becker DV. Radioiodine and the treatment of hyperthyroidism: the early history. Thyroid 1997;7:163-176. [ Links ]
127. Leslie WD, Ward L, Salamon EA, Ludwig S, Rowe RC, Cowden EA. A randomized comparison of radioiodine dose in Graves' hyperthyroidism. J Clin Endocrinol Metab 2003;88:978-983. [ Links ]
128. Crooks J, Buchanan WW, Wayne EJ, MacDonald E. Effect of pretreatment with methylthiouracil on results of 131I therapy. Br Med J 1960;1:151-154. [ Links ]
129. Tuttle RM, Patience T, Budd S. Treatment with propylthiouracil before radioactive iodine is associated with a higher treatment failure rate than therapy with radioactive iodine alone in Graves' disease. Thyroid 1995;5:243-247. [ Links ]
130. Berens S, Bernstein R, Robbins J, Wolff J. Antithyroid effects of lithium. J Clin Invest 1970;49:1357-1367. [ Links ]
131. Briere J, Pousset G, Darsy et Guinet P. The advantage of lithium in association with 131I in the treatment of functioning metastasis of the thyroid cancer. Ann Endocrinol 1974;35:281-282. [ Links ]
132. Gershengorn MC, Izumi M, Robbins J. Use of lithium as an adjunct to radioiodine therapy of thyroid carcinoma. J Clin Endocrinol Metab 1976;42:105-111. [ Links ]
133. Pons F, Carrio I, Estorch M, Ginjaume M, Pons J, Milian R. Lithium as an adjunct of iodine-131 uptake when treating patients with well-differentiated thyroid carcinoma. Clin Nucl Med 1987;12:644-647. [ Links ]
134. Bal CS, Kumar A, Chandra P. Effect of iopanoic acid on radioiodine therapy of hyperthyroidism: long-term outcome of a randomized controlled trial J. Clin Endocrinol Metab 2005;90:6536-6540. [ Links ]
135. Rubin L. Diagnosis and management of pulmonary artery hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004;126:7-10. [ Links ]
136. Arroliga A, Dweik R, Rafanan A. Primary pulmonary hypertension and thyroid disease. Letter to the editor. Chest 2000;118:1224 [ Links ]
137. Du L, Sullivan C, Chu D. Signaling molecules in nonfamilial pulmonary hypertension. N Engl J Med 2003;348:500-509. [ Links ]
138. Paolini R, Armigliato M, Zamboni S. Pulmonary hypertension in systemic diseases. Curr Drug Targets Inflamm Allergy 2004;3:459-467. [ Links ]
139. Vargas F, Rivas A, Osuna A. Effects of methimazole in the early and established phases of Ng-nitro-arginine methyl ester hypertension. Eur J Endocrinol 1996;135:506-513. [ Links ]
140. Marvisi M, Zambrelli P, Brianti M, et al. Pulmonary hypertension is frequent in hyperthyroidism and normalizes after therapy. Eur J Inter Med 2006;17:267-271. [ Links ]
141. Ardeshir S, Burstein S, Guy W, et al. Pulmonary hypertension in men with thyrotoxicosis. Respiration 2005;72;990-994. [ Links ]
142. Martí V, Ballester M, Rigla M, Narula J, Berna L, Pons-Llado G, Carrió I, Carreras F, Webb SM. Myocardial damage does not occur in untreated hyperthyroidism unless associated with congestive heart failure. Am Heart J 1997;134(6):1133-1137. [ Links ]
143. O'Leary DH, Polak JF, Kronmal RA, Manolio TA, Burke GL, Wolfson Jr SK. Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N Engl J Med 1999;340:14-22. [ Links ]
144. Chambless LE, Heiss G, Folsom AR, Rosamond W, Szklo M, Sharrett AR, Clegg LX. Association of coronary heart disease incidence with carotid arterial wall thickness and major risk factors: the Atherosclerosis Risk in Community (ARIC) Study, 1987-1993. Am J Epidemiol 1997;146:483-494. [ Links ]
154. Chambless LE, Folsom AR, Clegg LX, Sharrett AR, Shahar E, Nieto FJ, Rosamond WD, Evans G. Carotid wall thickness is predictive of incident clinical stroke: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Epidemiol 2000;151:478-487. [ Links ]
146. Völzke H, Robinson DM, Schminke U, Lüdemann J, Rettig R, Felix SB, et al. Thyroid function and carotid wall thickness. J Clin Endocrinol Metab 2004;89(5):2145-2149. [ Links ]
147. Garcia-Estan J, Atucha NM, Quesada T, Vargas F. Involvement of the renin-angiotensin system in the reduced pressure natriuresis response of hyperthyroid rats. Am J Physiol 1995;268:E897-E901. [ Links ]
148. Paul M, Ganten D. The molecular basis of cardiovascular hypertrophy. The role of the renin-angiotensin system. J Cardiovasc Pharmacol 1992;19:S51-S58. [ Links ]
149. Kato H, Suzuki H, Tajima S, Ogata Y, Tominaga T, Sato A, Saruta T. Angiotensin II stimulates collagen synthesis in cultured vascular smooth muscle cells. J Hypertens 1991;9:17-22. [ Links ]
150. Sernia C, Marchant C, Brown L, Hoey A. Cardiac angiotensin system receptors in experimental hyperthyroidism in dogs. Cardiovasc Res 1993;27:423-428. [ Links ]
151. Inaba M, Henmi Y, Kumeda Y, Ueda M, Nagata M, Emoto M, Ishikawa T, Ishimura E, Nishizawa Y. Increased stiffness in common carotid artery in hyperthyroid Graves' disease patients. Biomed Pharmacother 2002;56:241-246. [ Links ]
152. Czarkowski M, Hilgertner L, Powalowski T, Radomski D. The stiffness of the common carotid artery in patients with Graves' disease. Int Angiol 2002;21:152-157. [ Links ]
153. Glagov S, Vito R, Giddens DP, Zarins CK. Micro-architecture and composition of artery walls: relationships to location, diameter and the distribution of mechanical stress. J Hypertens 1992;10:S101-S104. [ Links ]
154. Squizzato A, Gerdes VEA, Brandjes DPM, Buller HR, Stam J. Thyroid diseases and cerebrovascular disease. Stroke 2005;36:2302-2310. [ Links ]
155. Hofbauer LC, Heufelder AE. Coagulation disorders in thyroid diseases. Eur J Endocrinol 1997;136:1-7. [ Links ]
156. Marongiu F, Cauli C, Mariotti S. Thyroid, hemostasis and thrombosis. J Endocrinol Invest 2004;27:1065-1071. [ Links ]
157. Shih CH, Chen SL, Yen CC, Huang YH, Chen CD, Lee YS, Lin KH. Thyroid hormone receptor dependent transcriptional regulation of fibrinogen and coagulation proteins. Endocrinology 2004;145:2804-2814. [ Links ]
158. Squizzato A, Romualdi E, Büller HR, Gerdes VEA. Thyroid dysfunction and effects on coagulation and fibrinolysis: a systematic review. J Clin Endocrinol Metab 2007;92:2415-2420. [ Links ]
159. Rogers II JS, Stanley RS. Factor VIII activity in normal volunteers receiving oral thyroid hormone. J Lab Clin Med 1983;102:444-449. [ Links ]
160. Awadi MH, Ho L, Dejong DC. Effect of TRH on plasma arginin vasopressin. Horm Res 1984;19:91-96. [ Links ]
161. Rem C. Blood coagulation, fibrinolytic activity and lipid profile in subclinical thyroid disease: subclinical hyperthyroidism increases plasma factor X activity. Clin Endocrinol (Oxf) 2006;64:323-329. [ Links ]
162. Fuster V, Rydén LE, Cannom DS, Crijns HJ, Curtis AB, et al. ACC/AHA/ESC 2006 Guidelines for the management of patients with atrial fibrillation. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 2006;114:e257-e354. [ Links ]
163. Umpierrez G, Challapalli S, Patterson C. Congestive heart failure due to reversible cardiomyopathy in patients with hyperthyroidism. AMJ Med Sci 1995;310:99-102. [ Links ]
164. Goldman L, Sahlas D, Sami M. A case of thyrotoxicosis and reversible systolic cardiac dysfunction. Can J Cardiol 1999;15:811-814. [ Links ]
165. Kantharia B, Richards H, Battaglia J. Reversible dilated cardiomyopathy: an unusual case of thyrotoxicosis. AM Heart J 1995;129:1030-1032. [ Links ]
166. Merce J, Ferras S, Oltra C, et al. Cardiovascular abnormalities in hyperthyroidism: a prospective Doppler echocardiographic study. Am J Med 2005;118:126-131. [ Links ]
167. Canaris G, Manowitz N, Mayor G, Ridgway E. The Colorado thyroid disease prevalence study. Arch Intern Med 2000;160:526-530. [ Links ]
168. Ganong WF. Fisiología médica. 16ª ed. México: Manual Moderno. 1998:355-372. [ Links ]
169. Froeschl M, Haddad H, Commons AS, Veinot JP. Thyrotoxicosis: an uncommon cause of heart failure. Cardiov Pathol 2005:14:24-27. [ Links ]

