










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.62 n.4 Ciudad Autónoma de Buenos Aires jul./ago. 2002
Evolución de la leucemia linfática crónica. Valor predictivo del inmunofenotipo, el CD23 soluble y la morfología
Marcela A. Sarmiento, María Fernanda Palacios, Mariano P. Scolnik, Federico R. Ramírez, Carmen Stanganelli, Juana Cabrera, Christian P. Chena, Guillermo Arrossagaray, Irma R. Slavutsky, Raquel M. Bengio
Departamentos de Clínica Hematológica, Inmunología Oncológica, Medicina Nuclear y Genética,
Instituto de Investigaciones Hematológicas, Academia Nacional de Medicina, Buenos Aires. Argentina.
Dirección postal: Dra. Marcela A. Sarmiento, Instituto de Investigaciones Hematológicas, Academia Nacional de Medicina, Pacheco de Melo 3081, 1425 Buenos Aires, ARGENTINA.
Fax: (54-11) 4803-9475. e-mail: msarmiento@hematologia.anm.edu.ar
Resumen
En la leucemia linfática crónica (LLC) se reconocen factores pronósticos útiles como la duplicación linfocitaria, el estadio clínico y el patrón de infiltración medular. Otros, como el porcentaje de células CD3+ están en estudio y requieren confirmación.
El objetivo del presente estudio fue evaluar si existe asociación entre morfología, inmunofenotipo linfocitario, CD23 soluble (SL) y sobrevida libre de eventos (SLE). Se evaluaron prospectivamente 36 pacientes sin tratamiento. Se determinaron: morfología típica, mixta y LLC-PL; inmunofenotipo linfocitario (score de Matutes); niveles plasmáticos de CD23 SL; estadio clínico, duplicación linfocitaria; b2microglobulina y alteraciones citogenéticas. Se consideró evento: progresión de enfermedad (necesidad de tratamiento, evolución a estadios avanzados, desarrollo de organomegalia voluminosa) y muerte por enfermedad. Mediana de seguimiento: 24 meses. Resultados: estadio 0: 11/36, SLE 80%; I: 10/36 SLE 90%; II: 13/36; III y IV: 2/36. SLE > II 37%. p= 0.023. Duplicación linfocitaria: <12m 7/31, >12m 24/31. SLE 28% vs 80% p<0.001. Citogenético: normal 13/28; anormal 15/28. SLE 92% vs 54%, p=0.053. +12 positiva 7/30, negativa 23/30. SLE 65% vs 66%. b2microglobulina normal 9/35, elevada 26/35. SLE 100% vs 53%, p=0.006. CD23 SL < 350 UI/ml 15/32, > 350 UI/ml 17/32. SLE 92% vs 53% p=0.005. Inmunofenotipo: Score 5: 15/36, Score 4: 19/36, SLE 64%. Score 3: 2/36. p=0.516. Morfología típica 17/35, mixta 17/35. SLE 81% vs 57%, p=0.099. LLC-PL 1/35. El CD 23 SL resultó adecuado para predecir SLE, particularmente útil en estadios iniciales sin otros marcadores de actividad. La morfología y el fenotipo linfocitario, dos variables accesibles, no fueron útiles para el propósito del estudio.
Palabras clave: Leucemia linfática crónica; Factores pronósticos.
Abstract
Evolution of chronic lymphocytic leukemia. Predictive value of immunophenotype, sCD23 and morphology. Some prognostic factors are useful in chronic lymphocytic leukemia (CLL): lymphocyte doubling time, clinical stage and bone marrow pattern infiltration, while others, such as the percentage of CD3+ cells, are under study and require confirmation. The objective of this study was to evaluate whether there is an association between morphology, lymphocyte immunophenotype, soluble CD23 (sCD23) and progression free survival (PFS). A total of 36 non-treated patients were enrolled. We analysed prospectively: morphology (typical, mixed and PL-CLL); immunophenotypic profile (Matutes score); sCD23 plasma levels; clinical stage; lymphocyte doubling time; b2microglobulin and karyotype abnormalities. Disease progression (need of treatment, progression to advanced stages, development of bulky organomegaly) and death related to disease were considered as events. Md of follow-up 24 mo. Results: Stage 0: 11/36, PFS 80%; I: 10/36 PFS 90%; II: 13/36; III and IV: 2/36. SLE > II PFS 37%. p= 0.023. Lymphocyte doubling time <12mo. 7/31; >12mo. 24/31. PFS 28% vs. 80% p<0.001. Karyotype: normal 13/28, abnormal 15/28. PFS 92% vs. 54% p=0.053. Trisomy 12: positive 7/30, negative 23/30, PFS 66% vs. 65%. b2 microglobulin: normal 9/35; high 26/35. PFS 100% vs. 53% p=0.006. sCD23 < 350 UI/ml: 15/32; > 350 UI/ml: 17/32. PFS 92% vs. 53% p=0.005. Immunophenotype: Score 5: 15/36, Score 4: 19/36, PFS 64%. Score 3: 2/36. p=0.516. Morphology: typical 17/35, mixed 17/35, PFS 81% vs. 57%, p=0.099. PL-CLL 1/35. Conclusions: sCD23 was suitable to predict PFS, specially useful for early stages without additional markers of active disease. Morphology (excluding PL-CLL) and immunophenotype, two common tools, were not useful for the study purpose.
Key words: Chronic lymphocytic leukemia; Prognosis factors.
La leucemia linfocítica crónica B (LLC-B) es un síndrome linfoproliferativo originado de la expansión neoplásica de una subpoblación de linfocitos de estirpe B, CD+. Dichas células se encuentran en reposo (G0) y presentan una sobrevida prolongada por inhibición patológica de la apoptosis, lo que conduce a su acumulación1-5. Este junto a otros mecanismos6-13 son en parte responsables de las características de la enfermedad.
La LLC-B tiene alta prevalencia en el mundo occidental, constituyendo el 30% de las leucemias del adulto, con una incidencia de 4/100 000 que aumenta a 50/100 000 en mayores de 70 años14.
La evolución clínica es variable. Se observan tanto formas indolentes con una expectativa de vida similar a la de la población sana del mismo grupo etario, como otras agresivas con una sobrevida aproximada de 2 años, generalmente asociada a enfermedad activa o avanzada15, 16. Numerosos investigadores analizaron la utilidad de parámetros clínicos y biológicos como factores pronósticos (FP) de evolución. Algunos han alcanzado aceptación universal; son ejemplos de ellos la edad, el patrón de infiltración en médula ósea (MO), la velocidad de duplicación linfocitaria en sangre periférica (SP), el estadio clínico (E) según clasificación de Rai o Binet17. Sin embargo otras características son objeto de estudio. Así, la morfología celular atípica y alteraciones citogenéticas como la trisomía 12, del 17p y 11q se relacionan con mal pronóstico18-20.
Marcadores serológicos como la ß2 microglobulina, componente del complejo mayor de histocompatibilidad, se postula como FP aun más importante que el estadio21, y la fracción soluble plasmática (SL) del CD23 parece ser útil en estadios tempranos de la enfermedad22-24. El inmunofenotipo linfocitario, si bien de valor diagnóstico25, como FP no mostró resultados concluyentes. Recientemente ha cobrado importancia la expresión del CD38 de membrana linfocitaria. Esta molécula es capaz de inducir apoptosis en linfocitos B inmaduros y rescata de la misma a los maduros26.
El estudio de las mutaciones de genes de inmuno-globulina VH26, 27 aporta evidencias de la existencia de dos tipos diferentes de linfocitos que darían origen a la LLC: linfocitos pregerminales y que por lo tanto no tuvieron oportunidad de experimentar mutaciones en dichos genes y linfocitos postgerminales que presentan mutaciones.
La importancia tanto del CD38 como de la mutación de los genes antes mencionados radica en que identifican dos subgrupos de pacientes con diferente evolución. Así, los casos de LLC que derivan de linfocitos pregerminales (no mutados) o que expresan en más de un 30% el CD38 presentan sobrevida más corta (< 7 años) y mayor agresividad que aquellos originados en linfocitos postgerminales (mutados) o con bajo porcentaje de células CD38+ quienes alcanzan sobrevidas cercanas a los 20 años. Cabe mencionar que pacientes con linfocitos no mutados presentan también una mayor ocurrencia de alteraciones genéticas desfavorables. Tanto las mutaciones Ig VH como la expresión del CD38 podrían manifestarse conjuntamente siendo parámetros independientes28.
Para que un FP alcance aceptación general debe tener valor independiente, sensibilidad, ser de fácil realización y bajo costo. Debe ser reproducido y confirmado antes de incorporarlo a la práctica médica. El análisis de los mismos permite identificar pacientes que evolucionarán desfavorablemente y considerar acertadas estrategias terapéuticas.
El objetivo del presente estudio fue evaluar si existe asociación entre la sobrevida libre de eventos (SLE) y la morfología celular, el inmunofenotipo de membrana linfocitaria y el CD23.
Materiales y métodosSe estudiaron prospectivamente 36 pacientes sin tratamiento previo, ingresados entre 1986 y 1999. Período de reclutamiento: 01/1997 a 06/1999. Las características de los pacientes y el período de seguimiento se muestran en la Tabla 1.

Al ingreso al protocolo se estadificaron mediante examen físico, hemograma, punción y biopsia de médula ósea, función hepática y renal, proteinograma electroforético, cuantificación de inmunoglobulinas, láctico deshidrogenasa (LDH), pruebas de Coombs directa e indirecta, orina completa, radiografía de tórax, tomografía computada y/o ecografía abdominal29.
Morfología celular: se evaluaron extendidos de SP (May Grunwald-Giemsa), realizando un recuento mayor a 100 linfocitos por 2 observadores. Se estableció según clasificación FAB morfología típica: presencia de linfocitos pequeños, de aspecto maduro con escaso citoplasma, núcleo con cromatina abigarrada y ausencia de nucléolos. Morfología atípica mixta: linfocitos con núcleos clivados o aspecto linfoplasmocitoide en más del 15% y LLC-PL: prolinfocitos entre un 10%-55% 30, 31.
Estadio clínico: según clasificación de RAI 16, 0: linfocitosis, I: linfocitosis y adenopatías, II: hepato o esplenomegalia, III: anemia y IV: plaquetopenia.
β2 microglobulina: se determinó por ELISA de competición (Inmunotech) en suero de pacientes y controles21. Las muestras y los estándares fueron incubados con un anticuerpo monoclonal en presencia de β2 microglobulina conjugada con fosfatasa alcalina. La actividad enzimática fue medida por sustrato cromogénico y la concentración fue obtenida por curva de estándares. Considerando los incrementos por edad, se definió como valor normal para < de 60 años: < 2.3 µg/ml y para > de 60 años un valor < 3 µg/ml.
CD23 soluble: se determinó en suero de pacientes y voluntarios sanos, por ELISA (Serotec, Oxford-England)32. Se incubaron 25 µL de suero en pocillos sensibilizados con anticuerpos anti-CD23 más anti-CD23 conjugado con peroxidasa, durante 2.30 hs. a temperatura ambiente. Se agregó el sustrato peróxido de hidrógeno y el cromógeno (3,3'5,5' tetrametil benzidina en base orgánica) por 20 minutos. Se estableció la curva estándar. Según testigos sanos se definió como valor normal 78 ± 14 DS UI/ml coincidente con el reportado en la literatura < 200 UI/mL22-24.
Inmunofenotipo linfocitario: se realizó en SP o MO por citometría de flujo con marcación de anticuerpos monoclonales con ficoeritrina e isotiocianato de fluoresceína. Se utilizaron anticuerpos para antígenos de superficie celular vinculados con patrones típicos y atípicos de LLC de acuerdo con el score diagnóstico de Matutes et al.25, 33 (Tabla 2), incluyendo cadenas livianas de inmunoglobulina (kappa/lambda, Kallestad, Austin, USA), CD5, CD19, CD20, CD22, CD23, FMC7, CD79b, y CD10 (Immunotech, Marseille, USA). Punto de corte de positividad del anticuerpo: 20% de la población B.

Estudio citogenético (CTG): se efectuó en SP y/o MO. Se realizó cultivo en medio F-10 suplementado con 15% de suero fetal bovino y estimulado con fitohemaglutinina (10 µg/ml) y pokeweed (20 µg/ml), durante 72 hs a 37°C, efectuándose el análisis cromosómico con técnica de bandeo G. Se complementó con la técnica de hibridación in situ con fluorescencia (FISH) para determinar la presencia de trisomía 12 y deleciones 13q y 17p. Se empleó la sonda de ADN a-satélite del cro-mosoma 12 (CEP 12, Spectrum Orange, VYSIS), procedién-dose de acuerdo a los protocolos establecidos. Por cada individuo se analizaron 400 núcleos interfásicos, estableciéndose como punto de corte para definir trisomía 12 la media de los controles (X±DS: 1.14 ± 0.56) más 3 desvíos estándar, y como criterio de positividad >2.82% de núcleos interfásicos con tres señales.
Eventos: signos de progresión de enfermedad (evolución a estadios más avanzados, necesidad de tratamiento específico, desarrollo de organomegalias voluminosas) y muerte por enfermedad.
Análisis estadístico: estudio de cohorte prospectivo. Nivel de significación de p < 0.05 con un poder de 80% e IC de 95%. Las variables numéricas continuas se analizaron por "t" de Student y las cualitativas por 2 y posteriormente regresión logística de Cox. La probabilidad de SLE se estimó por curvas de Kaplan-Meier y se compararon por Test de Log Rank.
Resultados
Estadio clínico: La distribución se muestra en Tabla 1. Para la evaluación de SLE los pacientes fueron agrupados en E0, EI y E > a II. La probabilidad de SLE a 24 meses fue del 80% para el E0, del 90% para el EI y del 37% para los E > II, p= 0.023.
Morfología celular: la población mostró en iguales porcentajes morfología típica y atípica mixta: 48.5% (17/35). Un paciente tuvo morfología LLC-PL y en un caso no fue determinada. Variedad atípica mixta: además de linfocitos con núcleo clivado y/o aspecto plasmocitoide, se observaron linfocitos con núcleo lobulado, otros de aspecto monocitoide, formas pequeñas con gránulos citoplasmáticos, formas inmaduras, otros con vellos polares y prolinfocitos en porcentajes menores al 10%. La probabilidad de SLE a 24 meses para la población con morfología típica fue del 81% y para la morfología mixta fue del 57%: p=0.099 (Fig. 1).

β2 microglobulina: la Md fue de 3.6 µg/ml (1-10µg/ml). El 74% (26/35) de la población presentó un valor anormal para su edad y el 26% (9/35) un valor normal. Un caso no fue determinado. Los valores anormales más altos predominaron en pacientes con masas voluminosas (Md 6.2 µg/ml, rango 3 a 10 µg/ml), morfología LLC-PL (10 µg/ml) y E avanzados: E 0: 2.6 µg/ml (1.8 a 5); E I: 2.2 µg/ml (1.2 a 8); E > II: 4.8 µg/ml (3.5 a 10). Para el análisis de SLE, los pacientes se agruparon según valores normales o anormales. La probabilidad de SLE a 24 meses para pacientes con valores normales fue del 100% y para ptes. con valores anormales fue del 53%: p=0.006.
Niveles plasmáticos de CD23 SL: 30/32 pacientes presentaron niveles elevados con una Md de 350 UI/mL. Pacientes en E0 presentaron valores altos en relación a estadios más avanzados: E0: Md 437.7 UI/ml (96 a 920), EI: Md 326 UI/ml (65 a 655) y E > II: Md 348 UI/ml (90 a 977). En ocho pacientes se repitieron las determinaciones al año y/o dos años de seguimiento, los valores permanecieron estables coincidiendo con la ausencia de progresión en todos excepto uno que mostró disminución luego del tratamiento.
Al evaluar los niveles de CD23 SL según el tiempo de evolución de la enfermedad, el 52% de los pacientes con enfermedad de menos de 1-2 años de evolución tuvieron valores elevados, coincidente con el 53% de los casos con más de 2 años de evolución.
Para el análisis de SLE los pacientes se agruparon según valores inferiores o superiores a la Md. La probabilidad de SLE a 24 meses para pacientes con valores inferiores fue del 92% y para aquellos con valores superiores fue del 53%, p = 0.005 (Fig. 2).

Inmunofenotipo linfocitario: score de 5 puntos: 15/36 ptes. (42%), 4 puntos: 19/36 (53%) y 3 puntos: 2/36 ptes (5%). El score de 4 puntos se debió principalmente a la alta prevalencia del FMC 7 en la población: 47%. Cinco pacientes no expresaron CD23 de membrana, tres de ellos presentaron eventos. Para la evaluación de la SLE se dividieron los pacientes en score de 4 y 5. La probabilidad de SLE a 24 meses de los grupos fue idéntica, 64%, p=0.516 (Fig. 3).

Estudio citogenético: por bandeo G se estudiaron 27 pacientes. Se realizó FISH para +12 en 30 pacientes y para las deleciones 13q y 17p en 24. Diez casos (37%) presentaron anomalías cromosómicas por citogenética convencional. De estos pacientes, seis mostraron deleción de parte del brazo largo del cromosoma 6, sola o acompañada por otras anomalías. Dos presentaron del(6)(q21), en un paciente como única alteración y el otro asociada a del(16)(q13), observándose progresión de la enfermedad en este último. Los 4 casos restantes presentaron del(6)(q15): en dos como única anomalía y con progresión, el tercero asociado a del(13)(q14q22) y el cuarto a inv(14)(q22q32) y con progresión de enfermedad. Otros 2 pacientes mostraron anomalías cromosómicas poco frecuentes en LLC: del(7)(q32) y trisomía 21, ambos sin progresión de enfermedad.
La trisomía 12 se observó en el 23% (7/30) de los casos: cinco presentaron cariotipo normal y +12 por FISH con valores entre 4.8 y 39%, un caso mostró +12 por citogenética y por FISH en porcentajes similares y el caso restante, previamente publicado34, mostró duplicación de parte del brazo largo del cromosoma 12 entre las bandas q13q22. Cinco de estos 7 casos presentaron progresión de la enfermedad.
La deleción 13q se encontró en 18/24, ocho de ellos progresaron (uno con trisomía 12 y otro con deleción 17p). La deleción 17 p se vio en un solo caso (negativo para +12 y positivo para del(13) que presentó mala evolución.
El análisis de SLE según tuvieran o no alteración citogenética, mostró una probabilidad de SLE a 24 meses del 92% para el grupo sin alteraciones citogenéticas y del 54% para el grupo con alteraciones, p=0.053. La +12 como variable única no influyó en la SLE: pacientes con +12 tuvieron una SLE del 65% y pacientes sin +12 del 66%, p= 0.338.
Duplicación linfocitaria: el 23% de los pacientes mostró una duplicación linfocitaria menor a 12 meses. La probabilidad de SLE a una Md de 24 meses fue del 80% para pacientes con duplicación mayor a 12 meses y 28% para menor a 12, p < 0.001 (Fig. 4).

Eventos: el 47% de la población (17/36) presentó eventos. Se observó evolución a estadios avanzados en seis casos, desarrollo de organomegalia voluminosa en siete, 10 ptes. necesitaron tratamiento y hubo 6 muertes por enfermedad. 9/17 pacientes mostraron morfología atípica y 8/17 alteraciones citogenéticas (+12 en cinco); 12/17 presentaron niveles basales elevados de CD23 SL y en 17 de b2 microglobulina con incrementos adicionales concomitantes al evento.
Tres pacientes fueron negativos para el CD23 por inmunofenotipo. Sólo 2 tuvieron LDH elevada y dos pertenecían a estadio de alto riesgo. El patrón de infiltración medular difuso fue visto en tres casos y porcentajes de infiltración > al 50% en nueve.
Ocho pacientes tuvieron alteraciones CTG siendo éstas una del(6)(q21),del(16)(q13); dos del(6)(q15); una del(6)(q15), inv(14), una del(17) y cinco trisomías del 12.
Al momento de este análisis, 6 pacientes han fallecido y más del 80% continúa vivo. (Tabla 3).
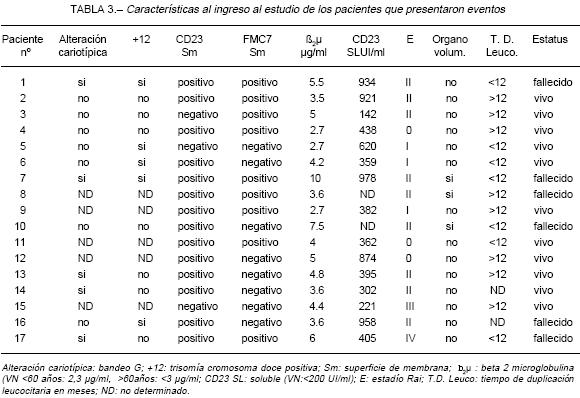
Alteración cariotípica: bandeo G; +12: trisomía cromosoma doce positiva; Sm: superficie de membrana; b2µ : beta 2 microglobulina (VN <60 años: 2,3 µg/ml, >60años: <3 µg/ml; CD23 SL: soluble (VN:<200 UI/ml); E: estadío Rai; T.D. Leuco: tiempo de duplicación leucocitaria en meses; ND: no determinado.
Discusión
El presente estudio tuvo como principal objetivo evaluar la utilidad pronóstica de tres diferentes variables. Dos de ellas, elementos de uso habitual para el diagnóstico de LLC: la morfología celular en SP (accesible y de bajo costo) y el inmunofenotipo linfocitario, y la tercera el CD23 soluble.
El análisis de la morfología linfocitaria mostró un alto porcentaje de casos atípicos mixtos: 47%, prevalencia superior a lo informado por otros investigadores (10%)19. Sólo un caso fue LLC-PL. Evaluando la importancia de la morfología para la aparición de eventos desfavorables, si bien la probabilidad de SLE a 24 meses fue mayor para la población típica (81%) que para la atípica (57%), la diferencia no fue estadísticamente significativa: p=0.099. Contrariamente Oscier et al19 encontraron que básicamente la LLC-PL pero también la mixta, tienen un alto riesgo de progresión de enfermedad en E0. Nuestra diferencia con respecto al mencionado autor se podría atribuir a la baja prevalencia de LLC-PL en nuestra casuística, a que nuestra población incluyó todos los estadios y finalmente, a que en el estudio de Oscier, las progresiones comienzan a verse claramente a partir del tercer año de seguimiento. En nuestros pacientes y dentro del período de seguimiento del estudio, la heterogeneidad morfológica no fue una información de valor predictivo de progresión de enfermedad. La morfología atípica, excluyendo de este análisis a la LLC-PL, no implicó peor sobrevida libre de eventos.
La evaluación del inmunofenotipo linfocitario se realizó teniendo en cuenta análisis previos del perfil antigénico típico para LLC presente en el 85-90% de los casos25, 33 y de la influencia adversa atribuida al FMC+ o al CD23 negativo35. Nuestra población presentó en un 47% expresión del antígeno FMC7, lo cual difiere con el 15% comunicado por otros autores35. La mayor prevalencia observada por nosotros podría deberse al corte determinado para positividad del marcador: 20% contra 30%. La ausencia del CD23 se vio en cinco casos y los eventos ocurrieron en tres que además presentaron FP adversos adicionales (b2 microglobulina alta, +12, acelerada duplicación leucocitaria, etc). En este punto nuestros resultados difieren de lo encontrado por Geisler et al35 cuyos pacientes CD23 negativos FMC+ tuvieron mala evolución. Pensamos que los eventos en nuestros tres pacientes pudieron atribuirse a otros FP más que al fenotipo expresado, ya que de nuestro estudio no surgen suficientes evidencias a favor de la influencia adversa atribuida a estos antígenos como únicas alteraciones. La identificación de estos últimos en nuestra población si bien fueron útiles en el contexto diagnóstico, no revelaron información adicional para predecir evolución.
Se evaluó la fracción soluble de CD23 para reproducir el dato sobre su valor predictivo en la evolución22-24. El hecho de escindirse de la superficie celular podría implicar algún evento biológico subyacente (actuaría como factor de crecimiento autocrino de células B al tiempo que impediría la instalación de la apoptosis). La técnica es sencilla y sus costos similares a los observados para b2 microglobulina. Mostró ser un marcador útil y un indicador precoz de la activación de la enfermedad. El mayor rendimiento clínico se obtuvo en pacientes con estadios iniciales en los que la ausencia de otros FP no permiten prever la evolución. El tiempo de evolución de la enfermedad no guardó relación con los niveles de CD23 SL. Creemos que su determinación es útil, especialmente en pacientes jóvenes quienes requieren una estrategia terapéutica individualizada.
Con respecto a FP ya aceptados, pudimos confirmar la utilidad de la b2 microglobulina, el estadio clínico y la duplicación leucocitaria, la cual mostró la diferencia de mayor significancia estadística (p < 0.001). La LDH y el patrón de infiltración medular no arrojaron información adicional.
Las alteraciones genéticas también evidencian que la LLC es una enfermedad heterogénea. El estudio citoge-nético de esta entidad por técnica de bandeo G permite detectar anomalías cromosómicas clonales en el 50% de los casos (35-36), valores que aumentan al 80-85% con la aplicación de la técnica de FISH20. Por medio de esta última se ha demostrado que las anomalías más comunes son, en orden de frecuencia, la deleción 13q14, deleción 11q22-23, la trisomía 12, la pérdida monoalélica de 17p13 (lugar donde se localiza el gen supresor de tumor p53) y la deleción 6q2120. Considerando separadamente a las anomalías numéricas, la trisomía 12 es la más frecuente, presente en un tercio de los pacientes estudiados con citogenética convencional36,37 y en el 12-35% de los casos analizados por FISH37-41 .
El análisis citogenético de nuestra población mostró en un 50% anomalías cromosómicas. La técnica de FISH detectó trisomía 12 en el 23% de los casos, del 13q en el 75% y del 17p en un solo un paciente, coincidiendo con otros autores39,40. Otra anomalía clonal detectada en nuestros pacientes fue la del(6q) con diferentes puntos de ruptura: del(6)(q15) y del(6)(q21). Se la observó en el 22% de los casos, este porcentaje es superior al 6-10% encontrado en la literatura20,42 . Respecto de esta alteración (frecuentemente responsable de la pérdida de material genético en neoplasias linfoides), las bandas más afectadas en LLC son 6q15 y 6q2343, situación observada en nuestra cohorte donde 4/6 pacientes mostraron el punto de ruptura en 6q15. El valor pronóstico de la trisomía 12 y la deleción 6q es controvertido20,36,41,45. Los pacientes con del(6q) no obstante presentar linfoadenopatías diseminadas y leucocitosis significativa, no tendrían evolución diferente respecto a los negativos para la delecion42. Se considera favorable la presencia de deleción 13q14 como única anomalía y las deleciones 17p y 11q conferirían mayor agresividad evolutiva y sobrevida más corta20. En nuestro estudio ocho pacientes con alteraciones cromosómicas experimentaron progresión: 5 con trisomía12, 4 con del(6q) y uno con del(17p). Cinco de ellos mostraron otras características que fueron asociadas a mala evolución (niveles elevados de CD23 SL y b2 microglobulina, acelerada duplicación leucocitaria) no obstante pertenecer a estadios no avanzados. La SLE fue similar en pacientes con y sin alteraciones citogenéticas. Para el período de seguimiento de este estudio con una Md de 24 meses, las alteraciones citogenéticas no fueron de valor pronóstico según los resultados estadísticos, aunque un mayor número de casos podría ponerlo de manifiesto. Dado la baja prevalencia de 17p en nuestra población, no podemos realizar conclusiones respecto de esta alteración. Cabe mencionar que no se evaluó deleción 11q. Es posible que la suma de factores adversos más que las alteraciones cariotípicas, expliquen la progresión de enfermedad en nuestros pacientes. Un mayor número de casos y un seguimiento más prolongado podría confirmar estos resultados. El análisis de SLE se realizó a 24 meses que es la mediana de seguimiento de la población estudiada, debido al bajo número de fallecimientos, no se realizó el cálculo de la sobrevida global.
Concluimos en primer término que en nuestra población, la heterogeneidad morfológica excluyendo la variante LLC-PL, la expresión del FMC7, la ausencia del CD23 de membrana, la trisomía 12 y la deleción 6q, no fueron factores adversos predictivos de eventos clínicos, durante el período de seguimiento considerado.
Concluimos finalmente, como aporte principal de este trabajo, que la fracción soluble del CD23 fue especialmente útil para pacientes en estadios tempranos que no mostraron otros FP asociados. Sugerimos su implemen-tación, particularmente en E < II que no evidencien otros parámetros de actividad, a fin de confirmar su aparente bajo riesgo. Teniendo en cuenta la biología de la LLC, un seguimiento más prolongado que permita observar la ocurrencia de un mayor número de eventos, permitirá corroborar los resultados obtenidos y analizar la probabilidad de sobrevida global.
En la actualidad, los interrogantes acerca de la diversidad del comportamiento clínico en LLC no están completamente aclarados. Consideramos que nuevos FP deben ser investigados y comparados con los reconocidos en modelos de probabilidad adecuados, concentrándose en estudiar aquellos que puedan superar en sensibilidad y ahorro de recursos a los actualmente aceptados. En tal sentido, futuros estudios aclararán la posibilidad de implementar en la clínica la información aportada por diversos moduladores de la respuesta biológica, como las IL4, IL6, IL 8 e IL10, el ICAM-1 y la timidina-kinasa entre otros17, de potencial utilidad como FP. Asimismo, combinar FP en un sistema de estadios de riesgo que contemple variables biológicas y clínicas podría optimizar el rendimiento de FP aislados.
Agradecimientos: Este estudio se realizó con el apoyo de la Fundación Alberto J. Roemmers y de la Secretaría de Ciencia y Técnica, subsidio PMT SID 0546.
1. Caligaris-Cappio F, Hamblin TJ. B-cell chronic lymphocytic leukemia: a bird of a different feather. J Clin Oncol 1999; 17: 399-401. [ Links ]
2. Rozman C, Montserrat E. Chronic lymphocytic leukemia. N Engl J Med 1995; 333: 1052-9. [ Links ]
3. Caligaris-Cappio F. B-chronic lymphocytic leukemia: a malignancy of anti-self B cells. Blood 1996; 87: 2615-20. [ Links ]
4. Stavisky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995; 268: 1749-52. [ Links ]
5. Wickremasinghe RG, Hoffbrand AV. Biochemical and genetic control of apoptosis: relevance to normal hemato-poiesis and hematological malignancies, Blood 1999; 93: 3587-90. [ Links ]
6. Inghirami G. Differential expression of LFA-1 molecules in non-Hodgkin's lymphoma and lymphoid leukemia. Blood 1988; 72: 1431-4. [ Links ]
7. Medeiros L J. Expression of LFA-1 in non-Hodgkin's lymphoma. Cancer 1989; 63: 255-7. [ Links ]
8. Strauder R. Expression of leukocyte function-associated antigen-1 and 7F7-antigen, an adhesion molecule related to intercellular adhesion molecule-1 (ICAM 1) in non-Hodgkin's lymphomas and leukemias: possible influence on growth patterns and leukemic behavior Clin. Exp. Imm. 1989; 77: 234-7. [ Links ]
9. Maio M. Differential expression of CD54 /intercellular adhesion molecule-1 in myeloid leukemias and in lympho-proliferative disorders. Blood 1990; 76: 783-5. [ Links ]
10. Pals S T. Expression of lymphocyte homing receptor as a mechanism of dissemination in non-Hodgkin's lymphoma. Blood 1989; 73: 885-9. [ Links ]
11. Picker L J. Expression of lymphocyte homing receptor antigen in non-Hodgkin's Lymphomas. Am. J. Pathol 1988; 130: 496-9. [ Links ]
12. Spertini, O. Regulation of leukocyte adhesion molecule-1 expression and shedding by normal and malignant cells. Leukemia 1991; 5: 300-4. [ Links ]
13. Tooze J. Decreased expression of complement receptor type 2 on neoplastic B cells of chronic lymphocytic leukaemia. Clin. Exp. Imm. 1991; 83: 423-3. [ Links ]
14. Caligaris-Cappio F. New insight into the biology of B-chronic lymphocytic leukemia. In: American Society of Hematology Education Program Book 1999, p 249-54. [ Links ]
15. Dameshek W. Chronic lymphocytic leukemia -an accu-mulative disease of immunologically incompetent lymphocytes. Blood 1967; 29: 566-70. [ Links ]
16. Rai KR, Sawitsky A, Cronkite EP et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975; 46: 219-22. [ Links ]
17. Molica S. Prognostic factors in chronic lymphocytic Leukemia. In: Cheson BD (ed). Chronic Lymphoid Leuke-mias. Second Edition, Revised and Expanded. Marcel Dekker, Inc New York Basel 2001: p 231-60. [ Links ]
18. Criel A, Verhoef G, Vlietinck R, et al. Further characteri-zation of morphologically defined typical and atypical CLL: a clinical, immunophenotypic, cytogenetic and prognostic study on 390 cases. B J Haematol 1997; 97: 383-91. [ Links ]
19. Oscier DG, Matutes E, Copplestone A, et al. Atypical lymphocyte morphology: an adverse prognostic factor for disease progression in stage A CLL independent of trisomy 12. B J Haematol 1997; 98: 934-9. [ Links ]
20. Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Eng J Med 2000; 343: 1910-6. [ Links ]
21. Keating, M J. The serum beta2-microglobulin level is more powerful than stage in predicting response and survival in chronic lymphocytic leukemia. Blood 1995; 86 (supp 1): 606a. [ Links ]
22. Sarfati M, Chevret S, Chastang C et al. Prognostic importance of serum soluble CD23 level in chronic lymphocytic leukemia. Blood 1996; 88: 4259-64. [ Links ]
23. Beguin Y, Lampertz S, De Groote D, Igot D, Malaise M, Fellet G. Soluble CD23 and other receptors (CD4, CD8, CD25; CD71) in serum of patients with chronic lympho-cytic leukemia. Leukemia 1993; 7: 2019-25. [ Links ]
24 Molica S, Levato D, Dell'Olio M, et. al. Cellular expression and serum circulating levels of CD23 in B-cell chronic lymphocytic leukemia. Implications for prognosis. Haema-tologica 1996; 81: 428-33. [ Links ]
25 Matutes E, Owusu-Ankomah K, Morilla R, Garcia Marco J, Houlihan A, Que T.H., Catovsky D. The immunologi-cal profile of B-cell disorders and proposal of a scor- ing system for the diagnosis of CLL. Leukemia 1994; 8: 1640-5. [ Links ]
26. Damble RN, Wasill T, Fais F, et al: Immunoglobulin V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999; 94: 1840-3. [ Links ]
27. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK: Unmutated immunoglobulin VH genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94: 1848-51. [ Links ]
28. Chiorazzi N, Ferrarini M. Immunoglobulin variable region gene characteristics and surface membrane phenotype define B-CLL subgroups with distinct clinical courses. In: Cheson BD (ed). Chronic Lymphoid Leukemias. Second Edition, Revised and Expanded. Marcel Dekker, Inc New York Basel 2001: p 81-109. [ Links ]
29. Cheson B, Bennett J, Grever M, Kay N, Keating M, O'Brien S, Rai K: National Cancer Institute-Sponsored Working Group Guidelines for chronic lymphocytic leukemia: Revised Guidelines for Diagnosis and Treatment. Blood 1996; 87: 4990-5.
30. Bennett JM; Catovsky D, Flandrin MT, Galton G, Gralnick DAG, Sultan C. Proposal for the classification of chronic (mature) B and T lymphoid leukaemias. J Clin Phat 1989; 42: 567-70. [ Links ]
31. D'Arena G, Keating M and Carotenuto M. Chronic lymphoproliferative disorders: an integrated point of view for the differential diagnosis. Leukemia and Lymphoma 2000; 36: 225-37. [ Links ]
32. Sarfati M, Bron D, Lagneaux L, Fonteyn C, Frost H, Delespesse G: Elevation of IgE-binding factors in serum of patients with B cell-derived chronic lymphocytic leukemia. Blood 1988; 71: 94-9. [ Links ]
33 .Moreau E, Matutes E, A'Hern R, Morilla R, et al. Improvement of the chronic lymphocytic leukemia scoring system with the monoclonal antibody SN8 (CD 79b). Hematopathology 1997; 108: 378-82. [ Links ]
34. Chena C, Sarmiento M, Palacios MF, Scolnik M, Slavutsky I. Dup(12)(q13-q22) and 13q14 deletion in a case of B-cell chronic lymphocytic leukemia. Acta Haematol 2000; 104: 197-201. [ Links ]
35. Geisler Ch, Larsen J, Hansen N, Hansen M, Christensen B, Lund B, et al. Prognostic importance of flow cytometric immunophenotyping of 540 consecutive patients with B-cell chronic lymphocytic leukemia. Blood 1991; 78: 1795-801. [ Links ]
36. Juliusson J, Oscier DG, Fitchett M, et al. Prognostic subgroups in B-cell chronic lymphocytic leukemia defined by specific chromosomal abnormalities. N Engl J Med 1990; 323: 720-5. [ Links ]
37. Oscier DG. Cytogenetic and molecular abnormalities in chronic lymphocytic leukemia Blood Rev 1994; 8: 88-97. [ Links ]
38. Perez-Losada A, Wessman M, Tiainen M et al. Trisomy 12 in chronic lymphocytic leukemia: An interphase cytogenetic study. Blood 1991; 78: 775-9. [ Links ]
39. Anastasi J, Le Beau MM, Vardiman JW, Fernald AA, Larson RA, Rowley JD. Detection of trisomy 12 in chronic lymphocytic leukemia by fluorescence in situ hybridization to interfase cells: A single and sensitive method. Blood 1992; 79: 1796-801. [ Links ]
40. Döhner H, Pohl S, Bulgay-Morschel M, Stilgenbauer S, Bentz M, Lichter P. Trisomy 12 in chronic lymphocytic leukemias - A metaphase and interphase cytogenetic analysis. Leukemia 1993; 7: 516-20. [ Links ]
41. Escudier SM, Pereira-Leahy JM, Drach JW et al. Fluores-cent in situ hybridization and cytogenetic studies of trisomy 12 in chronic lymphocytic leukemia. Blood 1993; 81: 2702-7. [ Links ]
42. Stilgenbauer S, Bullinger L, Benner A et al:. Incidence and clinical significance of 6q deletions in B cell chronic lymphocytic leukemia. Leukemia 1999; 13: 131-4. [ Links ]
43. Juliusson G, Oscier D, Gahrton G (IWCCLL). Cytogenetic findings and survival in B-cell chronic lymphocytic leukemia. Second IWCCLL compilation of data on 662 patients. Leukemia & Lymphoma 1991; 5: 21-5. [ Links ]
44. Bird ML, Ueshima Y, Rowley JD, Haren JM, Vardiman JW. Chromosome abnormalities in B-cell chronic lyumphocytic leukemia and their clinical correlations. Leukemia 1989; 3: 182-91. [ Links ]
45. Oscier DG, Stevens J, Hamblin TJ, Pickering RM, Lambert R, Fitchett M. Correlation of chromosome abnormalities with laboratory features and clinical course in B-cell chronic lymphocytic leukemia. B J Haematol 1990; 76: 352-8. [ Links ]
Recibido: 5-12-2001.
Aceptado: 17-07-2002.

