










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.63 n.4 Buenos Aires jul./ago. 2003
Embarazo luego de necrosis espontánea de un macroadenoma pituitario
Dobrovsky V., Garcia A. J., Artese R., Bruno O. D.
División Endocrinología, Hospital de Clínicas José de San Martín, Facultad de Medicina, Universidad de Buenos Aires
Resumen
La recuperación de la función pituitaria luego de necrosis de un macroadenoma es infrecuente pero no excepcional. Se presenta el caso de una mujer de 32 años de edad, previamente operada de una tumoración adrenal con signos sugestivos de hipercortisolismo que no revirtieron luego de cirugía. Más tarde presentó un cuadro de apoplejía pituitaria seguido de hipopituitarismo probado por mediciones hormonales. Una resonancia nuclear magnética (RNM) evidenció un macroadenoma pituitario con signos necróticos comprimiendo el quiasma óptico, realizándose su exéresis por vía transesfenoidal. Nueve meses después los estudios hormonales indicaron la casi total recuperación de la función pituitaria. La paciente tuvo un embarazo y parto normales tres años después. Luego de 5 años de seguimiento permanecía clínicamente asintomática, un test de hipoglucemia insulínica mostró reducción moderada en la respuesta del cortisol pero mayor en la de somatotrofina (GH) y una RMN demostró un aracnoidocele parcial, sin evidencia de tumor.
Palabras clave: Apoplejía pituitaria; Necrosis de macroadenoma pituitario.
Abstract
Although infrequent, recovery of pituitary function after necrosis of a pituitary adenoma is not an exceptional event. We report the case of a 32-year-old woman with previous surgery for an adrenal mass and signs of hypercortisolism which failed to revert postoperatively. She then developed pituitary apoplexy followed by hypopituitarism, as confirmed by hormonal measurements. Magnetic resonance imaging (MRI) showed evidence of a pituitary macroadenoma with signs of necrosis, impinging on the optic chiasm, which was excised by the trans-sphenoidal approach. Nine months later, hormone tests indicated a near total recovery of pituitary functions. The patient had a successful pregnancy three years later. After a 5-year follow-up, she remained clinically asymptomatic, with moderate reduction in cortisol and blunted growth hormone (GH) response to hypoglycemia and MRI failed to disclose any residual tumor, except for a partial arachnoidocele.
Key words: Pituitary apoplexy; Pituitary macroadenoma necrosis.
La necrosis espontánea de un macroadenoma pituitario puede acompañarse de remisión de un síndrome hiperfuncionante, con normalización de la función hipofisiaria1. Se comunica el caso de una paciente portadora de una aparente enfermedad de Cushing quien presentó, luego de necrosis espontánea de un macro-adenoma pituitario, signos de hipopituitarismo seguidos de embarazo y parto normales.
Caso clínico
Una mujer de 32 años consultó en otra institución en marzo de 1993 por signos sugestivos de sindrome de Cushing: hipertensión arterial, equimosis espontáneas, estrías violáceas, obesidad troncular, hirsutismo, amenorrea de 2 años de duración y un cortisol sérico basal de 39.8 µg/dl (vn 5.0-25.0). Una tomografía computada de abdomen superior demostró una masa adrenal derecha, que fue extirpada en junio de 1993; el examen anatomopatológico reveló un adenoma adrenocortical de 40 mm. Luego de cirugía, la hipertensión arterial desapareció pero los signos cushingoides persistieron; un cortisol sérico basal fue de 29.0 µg/dl, en febrero de 1994. Pocos días más tarde, la paciente debió ser asistida en un servicio de emergencia por presentar cefalea intensa, vómitos, trastornos visuales e hipotensión arterial, que revirtieron luego de la inyección intravenosa de glucocorticoides. Una resonancia nuclear magnética (RNM) de hipófisis mostró una masa de 25 x 25 mm con señal central de baja intensidad y compresión del quiasma óptico (Fig. 1). La campimetría visual computada puso en evidencia un escotoma nasal en ojo derecho. En el momento del episodio (marzo de 1994), los niveles basales de hormonas séricas eran sugestivos de hipopituitarismo: cortisol 2 µg/dl (vn 7-25), LH < 2 mIU/ml (vn 2-10), FSH 4 mIU/ml (vn 3-12), PRL 3 ng/ml (vn 5-20), T4 4 µg/dl (vn 4.5-12.5), T3 70 ng/dl (vn 70-210), TSH 2.9 µIU/ml (vn 0.3-5.0). La paciente fue enviada a nuestro hospital con el diagnóstico de apoplejía pituitaria, para exéresis quirúrgica de la masa. Al ingreso presentaba astenia, debilidad muscular, mialgias, irritabilidad, pérdida de memoria y había tenido una hemorragia de tipo menstrual luego de la apoplejía.

Al examen físico se constató un peso de 68 kg para una talla de 164 cm, con distribución centrípeta de la grasa corporal, facies redondeada y pletórica, hirsutismo, estrías abdominales y axilares pálidas, descamación de la piel en cara y atrofia muscular proximal; la tensión arterial era de 110/70 mmHg. Las determinaciones hormonales mostraron en ese momento valores subnormales de cortisol sérico y libre urinario (CLU) (6 µg/dl e indetectable, respectivamente; vn CLU: 20-90 µg/24 hs), respuesta normal de TSH al TRH (2.1 a 8.2 µIU/ml), T4 7.5 µg/dl (vn 4.5-12.5), T3 210 ng/ml (vn 70-210) y valores bajos de gonadotrofinas basales y post-LH-RH (LH < 0.5 a 0.9, FSH 0.3 a 3.0) con estradiol sérico elevado (259 pg/ml; vn 20-300). El diagnóstico de embarazo fue confirmado mediante la determinación de b-hCG en suero que arrojó un valor de 1000 mIU/ml, pero la paciente presentó un aborto espontáneo. El abordaje quirúrgico por vía transes-fenoidal permitió la remoción de una masa encapsulada líquida y amarillenta, informada por el patólogo como necrosis de un adenoma pituitario.
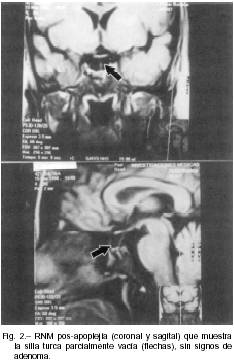
Cinco meses más tarde, la paciente aún exhibía niveles de cortisol sérico y urinario subnormales (3 µg/dl y 9 µg/24 hs, respectivamente), pero con normalización del test de LH-RH (LH 3.0 a 26.6, FSH 2.8 a 5.2); cuatro meses después, la investigación bioquímica indicaba recuperación funcional del eje hipófiso-adrenal (cortisol sérico 17 µg/dl, CLU 45 µg/24 hs). Desde entonces, la paciente continuó con menstrua-ciones regulares y obtuvo un nuevo embarazo con parto de un varón normal en febrero de 1998. Una RNM de hipófisis no evidenció signos de adenoma pero mostró un aracnoidocele parcial (Fig. 2). En ocasión de su última visita en mayo de 1998 lucía sana, no presentando signos cushingoides ni de insuficiencia anterohipofisiaria. En dicho momento, la evaluación hormonal mostró un CLU de 38.5 µg/24 hs con supresión normal del cortisol sérico matinal a las 8:00 hs (1.6 µg/dl luego de 1 mg de dexametasona, la víspera a las 23 hs); sin embargo, un test de hipoglucemia insulínica (glucemias: 81 a 19 mg/dl), demostró respuestas disminuidas de cortisol (12.1 a 18.8 mg/dl) y de somatotrofina (0.5 a 2.6 ng/ml).
Discusión
Esta paciente realizó su primera consulta por signos que, retrospectivamente, sugerian el diagnóstico de un síndrome de Cushing acompañado de hiperplasia macrono-dular secundaria, situación que puede observarse en hasta 10% de todos los casos de enfermedad de Cushing2. La eventual etiología del síndrome de Cushing no fue investigada ni antes ni después de la cirugía adrenal, aunque el cuadro clínico permanecía sin cambios. Luego de varios meses, la paciente desarrolló signos clínicos e imágenes de apoplejía pituitaria debidos a necrosis de un macroadenoma, confirmada por el estudio patológico de la pieza obtenida por cirugía transfenoidal.
La apoplejía pituitaria es un síndrome clínico caracterizado por el comienzo súbito de cefalea, trastornos visuales, vómitos y alteración del estado mental, causado por hemorragia o infarto de una glándula pituitaria normal o patológica1. En 1938, Sheehan y Murdoch3 describieron por primera vez el desarrollo de infarto pituitario luego de una hemorragia obstétrica. Aunque el infarto puede acaecer en una glándula aparentemente normal, ello sucede mucho más frecuentemente en un adenoma pre-existente4.
Wakai et al5 informaron sobre la frecuencia de apoplejía tumoral en una serie de 560 casos consecutivos operados en el transcurso de 30 años. Tuvieron 93 casos (16.1%), en los que la hemorragia en un adenoma pituitario fue confirmada, clínica o quirúrgicamente. Entre esos pacientes, 45.2% no presentaron síntomas vinculables con la hemorragia en tanto que el 54.8% restante sí fueron sintomáticos. El cuadro clínico de la apoplejía debe ser diferenciado de otros procesos intracraneanos agudos tales una hemorragia subaracnoidea, meningitis, infarto cerebral o trombosis del seno cavernoso.
La TC o la RNM son procedimientos por imágenes de gran utilidad para identificar hemorragia dentro de un adenoma pituitario1. Se ha descripto alta incidencia de necrosis hemorrágica en prolactinomas, somatotropinomas y adenomas no-funcionantes, sin que exista una prevalencia particular de alguno de esos subtipos5. Sólo existen comunicaciones aisladas de necrosis de micro o macro-corticotropinomas6, 7. Aunque en nuestro caso el tipo celular del adenoma no pudo ser demostrado dado su necrosis total, consideramos a partir de los datos clínico-bioquímicos que pudo haberse tratado de un macrocorticotropinoma. Hay pocos casos relatados en la literatura en que se demuestra por RNM la reducción de tamaño o desaparición del adenoma, con o sin una imágen de "silla turca vacía"8. Le Nestour et al6 informar sobre un paciente con remisión espontánea de enfermedad de Cushing luego de necrosis hemorrágica de un microadenoma verificado por RNM, con desaparición del mismo y aracnoidocele parcial un año más tarde.
Como consecuencia de una apoplejía puede desarrollarse una alteración transitoria o permanente de la función pituitaria9. El hipopituitarismo puede ser parcial o completo, según el grado de la necrosis pituitaria9, 10. La destrucción de menos de 40%-50% de la anterohipófisis se asocia rara vez con pérdida de función endocrina. Se requiere una destrucción mayor de 90% del parénquima glandular para el desarrollo de un panhipopituitarismo permanente. El hipopituitarismo puede deberse a la necrosis anterohipofisiaria o ser secundario a la disfunción hipotalámica inducida por la brusca expansión de la masa tumoral que provoca compresión de los vasos portales y/o del tallo pituitario11,12.
Pelkonen et al.10 estudiaron nueve pacientes que se habían recuperado espontáneamente luego de una apoplejía pituitaria, sin tratamiento quirúrgico. En todos los casos existió hipopituitarismo transitorio y cinco de ellos necesitaron terapia hormonal de reemplazo; dos de los cuatro restantes tenían enfermedad de Cushing y uno de ellos remitió luego de la apoplejía. Por otra parte, la función hipofisiaria alterada puede mejorar rápidamente luego del tratamiento quirúrgico. Baha et al.12 informaron ocho pacientes con apoplejía tumoral e hipopituitarismo tratados mediante cirugía descompresiva de urgencia. En todos ellos pudo documentarse la recuperación de la función glandular, independientemente de la extensión preoperatoria del déficit hormonal o de la duración de los síntomas. En esos casos, la mejoría en la función pituitaria fue atribuida al alivio del efecto de masa sobre la circulación portal y sobre el tallo pituitario. Por último, la recuperación tardía de la función pituitaria luego de apoplejía también está bien documentada. Martin13 comunicó el caso de un hombre de 35 años con remisión total de hipopituitarismo secundario a necrosis de un adenoma, 3 años después, probablemente explicable por replicación de células pituitarias normales remanentes. En nuestra paciente, cierto grado de hipopituitarismo pudo haber existido previamente a la apoplejía, como es habitual en los estados hipercortisólicos: la normalización de la función hipofisiaria luego de la apoplejía y cirugía subsecuente pudo resultar simplemente de la resolución del adenoma, ya que dicha normalización comenzó dentro de los primeros meses posteriores a la apoplejía, excepto para el eje adrenal que lo hizo a partir del quinto mes. La secreción de gonadotrofinas y ovulación recuperaron aparentemente en primer término, ya que luego de dos años en amenorrea, la paciente presentó menstruación y embarazo dentro de los dos meses pos-apoplejía. Aunque tuvo un aborto espontáneo, tres años más tarde cursó un embarazo con parto normal. Los estudios de control por imágenes demostraron un aracnoidocele parcial sin signos de residuo tumoral. Luego de un seguimiento de 5 años, la paciente sólo exhibía respuesta disminuida en cortisol y GH a la hipo-glucemia, configurando un hipopituitarismo parcial subclínico producido por la apoplejía o ligado a su probable enfermedad de Cushing previa.
1. Rolih C, Ober P. Pituitary apoplexy. Endocr Metab Clin North Am 1993; 22: 291-302. [ Links ]
2. Doppman M, Miller D, Dwyer M, et al. Macronodular adrenal hyperplasia in Cushing disease. Radiology 1988; 166: 347-52. [ Links ]
3. Sheehan H. & Murdoch R. Postpartum necrosis of the anterior pituitary: pathological and clinical aspects. J Obst Gynecol Br Empire 1938; 45: 456. [ Links ]
4. Randeva HS, Schoebel J, Byrne J, Esiri, Adams CB, Wass JA. Classical pituitary apoplexy: clinical features, management and outcome. Clin Endocrinol 1999; 51: 181-8. [ Links ]
5. Wakai S, Fukushima T, Teramoto A, Sano K. Pituitary apoplexy: its incidence and clinical significance. J Neurosurg 1981; 55: 187-93. [ Links ]
6. Le Nestour E, Bécassis J, Bertagna X, Bonnin A, Luton J. Silent necrosis of a pituitary corticotroph adenoma revealed by timely magnetic resonance imaging: a cause of spontaneous remission of Cushings disease. Europ J Endocrinol 1994; 130: 469-71. [ Links ]
7. Lazaro CM, Guo WY, Sami M, et al. Hemorrhagic pituitary tumours. Neuroradiology 1994; 36: 111-4. [ Links ]
8. Robinson DB, Michaels RD. Empty sella resulting from the spontaneous resolution of a pituitary macroadenoma. Arch Intern Med 1992; 152: 1920-3. [ Links ]
9. Pelkomen R, Kuusisto A, Salmi J et al. Pituitary function after pituitary apoplexy. Am J Med 1978; 65: 773-8. [ Links ]
10. Kovacs K. Necrosis of anterior pituitary in humans. Neuroendocrinology 1969; 4:170-9. [ Links ]
11. Veldhuis J, Hammond J. Endocrine function after spontaneous infarction of the human pituitary: report, review, and reappraisal. Endoc Rev 1980; 1: 100-7. [ Links ]
12. Baha M, Arafah J, Harrington F, Madhoun Z, Selman W. Improvement of pituitary function after surgical decompression for pituitary tumor apoplexy. J Clin Endocrinol Metab 1990; 71: 323-8. [ Links ]
13. Martin F. Spontaneous cure of hypopituitarism. The Endocrinologist 1994; 4: 184-8. [ Links ]
Recibido: 11 de marzo de 2003
Aceptado: 10 de junio de 2003

