










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680On-line version ISSN 1669-9106
Medicina (B. Aires) vol.63 no.6 Buenos Aires Nov./Dec. 2003
Leptina en pacientes con síndrome de ovario poliquístico. Correlación directa con la resistencia insulínica
Calvar C. E.1, Intebi A. D.1, Bengolea S. V.1, Hermes R.1, Spinedi E.2
1División Endocrinología, Hospital J. Fernández, Buenos Aires;
2Unidad de Neuroendocrinología, IMBICE, Universidad Nacional de La Plata
Resumen
La presencia de resistencia insulínica (RI) es un hecho frecuentemente asociado al síndrome de ovario poliquístico (PCO), sin embargo aún no está clara la relación existente entre la RI y los niveles circulantes de leptina en estas pacientes. En este trabajo investigamos los niveles plasmáticos de leptina en pacientes con PCO y evaluamos su relación con la presencia de RI. Se seleccionaron 49 pacientes con PCO y 14 mujeres normales. A todas las pacientes se les realizó un test de tolerancia oral a la glucosa. Se dosó leptina (LEP), insulina y glucosa séricas durante las 2 hs del test y se determinaron los índices de sensibilidad insulínica y relación leptina: índice de masa corporal (LEP:BMI). 1) No observamos diferencias significativas en los niveles séricos de LEP (ng/ml) entre las pacientes PCO (21.9 ± 2.8) y los controles normales (17.6 ± 4.9). 2) En las pacientes con PCO, los niveles séricos de LEP y el índice LEP:BMI se correlacionaron en forma significativa con el BMI y los índices de sensibilidad insulínica (P<0.01). 3) Diecisiete pacientes con PCO que presentaron RI evidenciaron niveles significativamente mayores de leptina sérica (32.8 ± 4.3 vs. 16.2 ± 3.2, P<0.01) y LEP:BMI (0.95 ± 0.09 vs. 0.61 ± 0.08, P<0.05) que las pacientes sin RI. En conclusión, nuestros resultados evidencian que el síndrome PCO parecería cursar con normoleptinemia, sin embargo la presencia de RI podría estar relacionada con un aumento de la concentración de leptina sérica independientemente del BMI. Estos resultados avalan la existencia de una interrelación leptina – insulina en este grupo de pacientes.
Palabras clave: Leptina; Resistencia insulínica; Síndrome de ovario poliquístico.
Abstract
Up to now it is unclear whether there is a relationship between insulin resistance and circulating leptin levels (LEP) in women with polycystic ovary syndrome (PCOS). To assess the role of LEP in PCOS and to clarify the relationship between plasma LEP levels and insulin resistance (IR) in PCOS patients, we studied 49 women with PCOS and 14 normal premenopausal women. All subjects were evaluated by a 2 hours, 75 g, oral glucose tolerance test. Fasting plasma LEP, insulin, glucose, insulin sensitivity indexes and LEP:body mass index (BMI) were determined. Results were analyzed by ANOVA and the Pearsons correlation test when appropriate. The results indicate that: 1) no differences were found in basal plasma LEP levels (ng/ml) between normal (17.6 ± 4.9) and PCOS (21.9 ± 2.8) women; 2) in PCOS patients, a significant (P < 0.01) correlation between plasma LEP levels and BMI and insulin sensitivity indexes were found; and 3) seventeen PCOS patients were insulin resistant (IR) and showed higher basal plasma LEP levels (32.8 ± 4.3, P < 0.01) and LEP:BMI (0.95 ± 0.09, P < 0.05) than non insulin resistant (non IR) PCOS subjects (16.2 ± 3.2 and 0.61 ± 0.08, respectively). Our results suggest that PCOS seems to be associated with normoleptinemia, however, if IR are analyzed separately from non IR PCOS patients, there is a clear relationship between IR PCOS and hyperleptinemia, regardless of the BMI. The present study strongly supports bi-directional relationship between fat and carbohydrate metabolisms under a very particular physiopathological condition (PCOS).
Key words: Leptin; Insulin resistance; Polycistic ovary syndrome.
La poliquistosis de ovario (PCO) es una de las endocrinopatías más comunes en pacientes en edad reproductiva, alcanzando una prevalencia entre el 5 y 10% en el grupo de mujeres premenopáusicas1. Se caracteriza por la presencia de hiperandrogenismo, anovulación crónica y/o ovarios multiquísticos. Clínica-mente las pacientes presentan en un alto porcentaje de los casos obesidad, hirsutismo y acné, trastornos del ciclo menstrual e infertilidad y en algunos otros virilización.
Con alta incidencia, se ha observado una asociación entre PCO y resistencia insulínica (RI) independientemente de la obesidad2-4 . La resistencia insulínica es uno de los factores patogénicos de la PCO, ya que la hiperinsulinemia puede ser causa de hiperandrogenismo actuando sobre el receptor de IGF 1 de las células de la teca estimulando la síntesis de andrógenos en forma sinérgica con la hormona luteinizante (LH)5-7.
Debido a la alteración en la acción de la insulina, las pacientes portadoras de PCO presentan un riesgo aumentado de contraer diabetes no insulino dependiente. La prevalencia de intolerancia a la glucosa en mujeres premenopáusicas es del 5.3%8 y en mujeres obesas con PCO alcanza el 20%. Teniendo en cuenta que el 50 a 60% de las mujeres portadoras de PCO son obesas1, 9, la PCO contribuiría al 10% de los casos de intolerancia a la glucosa en mujeres premenopáusicas.
La leptina, el producto del gen de la obesidad (ob), es sintetizada en el tejido adiposo10. Esta proteína regula los depósitos de grasa y de esta forma el peso corporal, afectando el apetito y la termogénesis11. Los niveles plasmáticos de leptina se encuentran aumentados en varios modelos de obesidad animal así como en la obesidad humana, y se correlacionan en forma positiva con el grado de obesidad12. Por otra parte, se ha propuesto que la leptina serviría como una señal permisiva de las funciones reproductivas13.
Los niveles plasmáticos de insulina en ayunas se correlacionan con los niveles circulantes de leptina que se encuentran aumentados en algunos pacientes con RI14-16. Sin embargo, el rol de la insulina en la regulación de los niveles circulantes de leptina es aún controvertido17-19.
Las mujeres portadoras de PCO sirven como modelo para evaluar las interacciones existentes entre RI e hiperinsulinemia crónica con los niveles séricos de leptina, debido a la alta frecuencia de resistencia insulínica e hiperinsulinemia en asociación con obesidad, que las mismas presentan2, 3. Sin embargo, actualmente se desconoce el preciso rol de la leptina en el síndrome PCO20. Se ha informado que las mujeres portadoras de PCO pueden presentar niveles plasmáticos de leptina aumentados21, 22 o normales23-27, en relación con los controles sanos. Asimismo, existen controversias acerca de los cambios en los niveles plasmáticos de leptina en respuesta a diferentes terapias insulino sensibilizantes. El tratamiento de pacientes portadoras de PCO con troglitazona no modificó en forma significativa los niveles plasmáticos de leptina a pesar de disminuir significativamente las concentraciones séricas de insulina24. Sin embargo, en estudios recientes26-28, se observó una disminución significativa de los niveles plasmáticos de leptina e insulina, sin cambios en el índice de masa corporal, en mujeres con PCO tratadas con metformina o diazóxido. Estos estudios sugieren que la disminución de los niveles de insulina podría jugar algún rol en la disminución de las concentraciones séricas de leptina. Los objetivos del presente trabajo fueron: a) evaluar el rol de la leptina en el síndrome de PCO y b) clarificar la relación existente entre los niveles plasmáticos de leptina y la resistencia insulínica en las pacientes con PCO.
Materiales y métodos
Pacientes
Se estudiaron 49 pacientes portadoras de PCO (rango de edad: entre 17 y 42 años) y 14 mujeres normales (rango de edad: entre 17 y 42 años); las mujeres de este último grupo (grupo control) reunieron los siguientes criterios: ciclos menstruales regulares, sin evidencias de hirsutismo, hiperandrogenemia o historia familiar de diabetes. Todas las pacientes se encontraban eutiroideas, normoprolactinémicas y ninguna había recibido terapia farmacológica alguna durante los tres meses previos a la iniciación del estudio. El diagnóstico de PCO se realizó por la presencia de a) amenorrea u oligomenorrea persistente de inicio puberal asociado a hirsutismo y/o acné, b) aumento de los niveles séricos de testosterona y/o delta-4-androstenediona y c) evidencia ecográfica de ovarios aumentados de tamaño y poliquísticos. Se excluyeron las pacientes portadoras de hiperplasia suprarrenal congénita no clásica o síndrome de Cushing mediante el uso de los estudios apropiados.
Protocolo de estudio
Todos los sujetos fueron estudiados entre los días 3 a 8 del ciclo menstrual o en amenorrea (pacientes sin menstruación por más de 90 días), para la determinación de hormona luteinizante (LH) y hormona folículo estimulante (FSH) plasmáticas (ELISA) y testosterona y estradiol plasmáticos (RIA).
A todas las mujeres se les realizó el test de tolerancia oral a la glucosa (TTOG). Luego de la inserción de un catéter endovenoso, se obtuvieron muestras de sangre antes de la ingesta (tiempo cero) y 30, 60, 90, 120, 150 y 180 minutos después de la ingesta de una carga oral de 75 g de glucosa. Las muestras fueron utilizadas para la determinación de las concentraciones séricas de glucosa (hasta los 120 minutos) (método enzimático), insulina (hasta los 120 minutos) (MEIA; Abbott) y leptina (hasta los 180 minutos) (IRMA, Diagnostic System Lab).
La valoración antropométrica se realizó mediante el índice de masa corporal (BMI): peso/talla2.
La distribución de grasa se evaluó mediante el índice de cintura/cadera (C:C). La circunferencia de cintura se midió en la mitad de la distancia entre la última costilla y la cresta ilíaca y la circunferencia de cadera se midió como la máxima circunferencia sobre los glúteos (ambas como la media de 3 mediciones consecutivas)29. El estudio fue aprobado por el comité de ética.
Análisis de los datos
Se determinó el índice glucemia/insulinemia (GLU:INS) de las concentraciones de glucosa e insulina basales, el modelo homeostático (HOMA)30, el índice de sensibilidad insulínica (ISI)31 y el índice leptina/BMI (LEP:BMI). Se calculó el área bajo la curva de las concentraciones de insulina (AI) y glucosa (AG) durante el TTOG, por el método trapezoidal32.
El diagnóstico de resistencia insulínica se realizó por la presencia de por lo menos tres de los siguientes criterios: insulinemia en ayunas > a 16.9 µUI/ml, AI > 11420, GLU:INS < 4.2, ISI < 3.3 y modelo homeostático (HOMA) > 3.2.
Los resultados, expresados como la media ± error estándar (ES), fueron analizados por ANOVA (seguido del test de Fisher) y por el test de Student (para comparación de las medias) y el test de correlación de Pearson, cuando fue necesario.
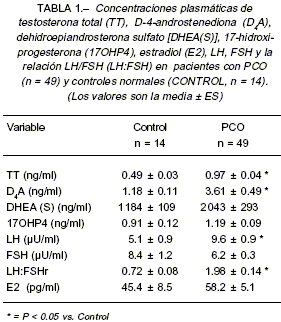
Resultados
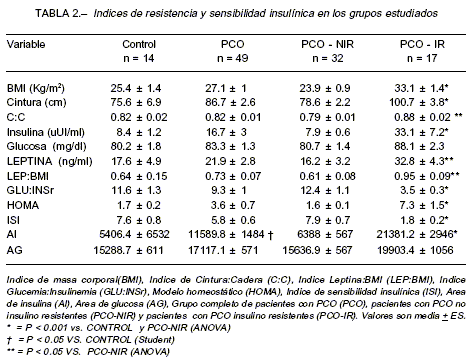
Los datos de los resultados de laboratorio endocrino-lógico se muestran en la Tabla 1. Las pacientes con PCO presentaron niveles plasmáticos de insulina en ayunas mayores (16.7 ± 3 µUI/ml) que los controles normales (8.4 ± 1.2 µUI/ml), sin embargo las diferencias no fueron estadísticamente significativas. Como puede verse en la Tabla 2, se observó una mayor AI en las pacientes PCO comparadas con el grupo control (11.589.8 ± 1.484 vs. 5.406.4 ± 6.532; P < 0.05; Fig. 1).

No hubo diferencias significativas en los niveles de leptina plasmática (LEP) en ayunas entre los controles (17.6 ± 4.9 ng/ml) y las pacientes con PCO (21.9 ± 2.8 ng/ml). (Fig. 2).

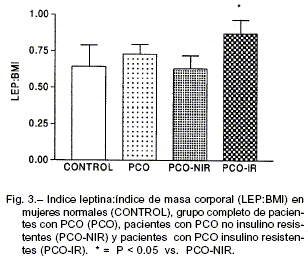
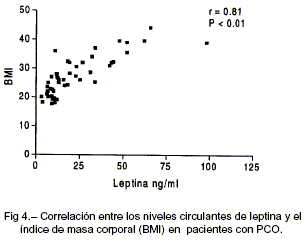
Diecisiete mujeres con PCO presentaron resistencia insulínica (PCO-IR) respecto de las normales. Como puede verse en la Tabla 2, en estas pacientes todos los índices de sensibilidad insulínica fueron significativa-mente menores, así como los índices de resistencia insulínica significativamente mayores respecto de los controles normales y de las pacientes PCO sin RI (PCO-NIR). Los niveles circulantes de LEP (32.8 ± 4.3 ng/ml) fueron significativamente mayores (P < 0.05) que en las pacientes PCO-NIR (16.2 ± 3.2 ng/ml; Fig. 2). Dado que las pacientes PCO-IR también presentaron un BMI significativamente mayor que el grupo PCO-NIR, para evitar el efecto del mismo sobre la diferencia en los niveles plasmáticos de LEP, evaluamos el índice LEP:BMI y pudimos observar que el índice LEP:BMI fue significativamente mayor (P < 0.05) en las pacientes PCO-IR (0.95 + 0.09) que las pacientes PCO-NIR (0.61 + 0.08; Fig. 3). En el grupo de pacientes con PCO observamos una correlación significativa (P<0.01) entre los niveles plasmáticos de LEP y el BMI (r=0.81) (Fig. 4); el índice GLU:INS (r=-0.46) y el ISI (r=0.51); así como entre los índices LEP:BMI y GLU:INS (r=-0.45) y el ISI (r=-0.50) (P<0.01).
Con respecto a los valores antropométricos, pudimos observar una correlación positiva (P<0.01) entre los niveles plasmáticos de LEP y el índice cintura/cadera (C:C) (r=0.42) así como entre el índice LEP:BMI y el índice C:C (r=0.36; P<0.05), en el grupo de pacientes PCO.
No hallamos correlaciones significativas entre los niveles plasmáticos de leptina y los valores de testosterona, estradiol, LH y FSH séricos.
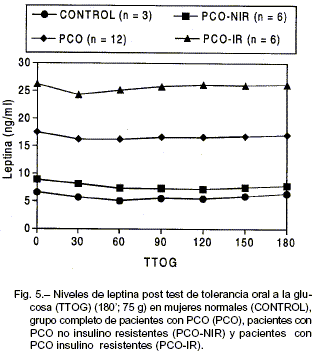
Durante el TTOG las concentraciones de leptina permanecieron sin cambios a pesar del aumento de los niveles de glucosa e insulina (Fig. 5).
Discusión
En nuestro grupo de pacientes con PCO pudimos observar niveles de leptina plasmática similares a los valores de un grupo de mujeres sanas con ciclos ovulatorios y con un BMI comparable, así como una correlación positiva entre leptina circulante y BMI, como ha sido previamente informado23-25, 33. Sin embargo, un sub-grupo de pacientes con PCO portadoras de resistencia insulínica, presentó niveles elevados de leptina, independientemente de su BMI. Este último resultado, asociado al hallazgo de correlaciones significativas entre los niveles de leptina y diferentes parámetros de hiperinsulinemia, sugieren la presencia de una interrelación entre la resistencia insulínica y los niveles de leptina en nuestras pacientes portadoras de PCO.
La PCO es una entidad claramente asociada a la presencia de hiperinsulinemia y RI4,5,7. Un gran número de genes en diferentes vías metabólicas han sido propuestos como posibles genes susceptibles relacionados con la PCO34. Aparentemente, los genes susceptibles para PCO estarían localizados en el cromosoma 19p13.3, en la región del gen del receptor de insulina35. La resistina es un polipéptido secretado por el tejido adiposo que modularía la acción de la insulina y la tolerancia a la glucosa36, 37. Se ha postulado que la resistina podría estar involucrada en la resistencia insulínica relacionada con la PCO; sin embargo, en un estudio reciente se observó una baja probabilidad de asociación entre las variaciones del gen de la resistina y el fenotipo de PCO en varias familias estudiadas38.
Varias publicaciones confirman un rol regulador de insulina sobre la producción de leptina tanto en animales como en humanos39-43. De esta forma, la insulina parecería ser capaz de regular la expresión de leptina en diferentes modelos de roedores y, luego de una infusión prolongada, en el hombre39-42. Adicionalmente, datos encontrados "in vivo" sugieren que la expresión de ARN mensajero de leptina en el tejido adiposo de ratas puede ser estimulado por infusiones cortas de insulina43, hiperinsulinemia post prandial y una inyección aislada de insulina40. En estudios recientes se ha observado una disminución de los niveles plasmáticos de leptina en asociación a una disminución de la insulinemia en pacientes con PCO tratadas con metformina y diazóxido (agentes insulino-sensibilizadores)26-28. Probablemente la disminución de los niveles plasmáticos de leptina en estas investigaciones fueron el resultado de la disminución de insulina, ya que las pacientes no variaron su BMI luego de la terapia; estos hallazgos sugerirían una importante interrelación metabólica entre leptina e insulina en pacientes con PCO. En contraposición con estos resultados mencionados, el tratamiento con troglitazona en pacientes con PCO, no indujo una disminución de los niveles de leptina a pesar de la reducción de los niveles plasmáticos de insulina24. Las discrepancias en los resultados podrían ser explicadas en parte por las diferencias en el/los mecanismo/s de acción de las drogas. Es interesante destacar el efecto inhibidor de la síntesis de leptina que ha sido atribuido a la troglitazona44.
Brzechffa y col. observaron niveles séricos elevados de leptina en un sub-grupo de pacientes con PCO al compararlas con mujeres sanas con ciclos regulares21. La elevación de los niveles plasmáticos de leptina, en las pacientes publicadas por Brzechffa, no estaba relacionada con la hiperinsulinemia como sí lo observamos en nuestro grupo de pacientes con PCO. Desconocemos la causa exacta de las diferencias entre estos resultados, sin embargo, en parte al menos, podrían ser explicadas por diferencias étnicas y/o de alimentación entre las dos poblaciones estudiadas.
La interacción de los niveles circulantes de leptina con los de gonadotrofinas y esteroides gonadales es de particular interés. Varias evidencias sugieren que la leptina sería una señal metabólica que jugaría un rol importante en la regulación del eje hipotálamo-hipófiso-gonadal45-48. Se ha observado que el tratamiento crónico con leptina además de reducir el peso corporal y mejorar la resistencia insulínica, corrige el hipogonadismo del ratón ob/ob13, 45. Además, el tratamiento con leptina acelera la pubertad del ratón normal46 y el seguimiento longitudinal de los niveles de leptina durante la pubertad en niños revela que la misma aumenta inmediatamente antes del inicio de la pubertad y ha sido sugerido que este evento podría ser la señal metabólica para el inicio de la pubertad en humanos47, 49. Debido a que el PCO es un síndrome de patogenia aún no del todo clara, que cursa con una disfunción del eje hipotálamo-hipófiso-ovárico1, y que la leptina jugaría un rol importante en la regulación del eje gonadal45, 48, las modificaciones en los niveles plasmáticos de leptina, como se observa en algunas pacientes con PCO, podrían tener alguna implicancia en las alteraciones que se observan en los diferentes niveles de regulación de la liberación de esteroides gonadales, o bien por el contrario, ser un mecanismo compensador tendiente a regular el eje hipotálamo-hipófiso-gonadal. Nuestros resultados no avalan la presencia de una relación directa entre las concentraciones circulantes de leptina y la liberación de LH/ FSH ni con la de esteroides gonadales, en pacientes con PCO, observación que encuentra coincidencia con resultados divulgados en otras investigaciones previas21, 23-25.
En el presente estudio encontramos que la leptina se mantuvo estable en los diferentes tiempos durante el TTOG. Hasta el momento, y de acuerdo a nuestro conocimiento, esta sería la primera investigación que ha evaluado este hecho en pacientes con PCO. Según nuestros resultados, la ausencia de respuesta aguda de leptina circulante post-hiperinsulinemia postprandial en PCO sería similar a lo informado en sujetos sanos delgados, en obesos y en pacientes diabéticos14, 17, 42, 50.
En conclusión, la PCO parecería estar asociada a la presencia de normoleptinemia, sin embargo al evaluar a las pacientes PCO con resistencia insulínica (RI) como un sub-grupo, pudimos observar una clara relación entre la presencia de RI y el aumento de los niveles plasmáticos de leptina independientemente del BMI. Nuestra investigación sugiere la presencia de una relación bi-direccional entre el metabolismo de lípidos e hidratos de carbono en una situación fisiopatológica particular como es el síndrome de PCO. Durante el TTOG, los niveles de leptina no se modificaron a pesar del aumento en los niveles de insulina plasmática. Según nuestro conocimiento, éste es el primer estudio en evaluar este hecho.
1. Dunaif A, Givens JR, Haseltine F, Merriam GR. The Poly-cystic Ovary Syndrome. Cambridge: Blackwell Scientific 1992. [ Links ]
2. Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance independent of obesity, in polycystic ovary syndrome. Diabetes 1989;38:1165-73. [ Links ]
3. Dunaif A. Hyperandrogenic anovulation (PCOS): A unique disorder of insulin action associated with an increased risk of non-insulin-dependent diabetes mellitus. Am J Med 1995; 98: 33-9. [ Links ]
4. Dunaif A, Wu X, Lee A, Diamanti-Kandarakis E. Defects in insulin receptor signaling in vivo in the polycystic ovary syndrome (PCOS). Am J Physiol Endocrinol Metab 2001; 281: E392-E399. [ Links ]
5. Barbieri RL, Smith S, Ryan KJ. The role of hyperinsulinemia in the pathogenesis of ovarian hyperandrogenism. Fertil Steril 1988; 50: 197-212. [ Links ]
6. Nobels F, Dewaiily D. Puberty and polycystic ovary syndrome: The insulin/insulin-like growth factor hypothesis. Fertil Steril 1992; 58: 655-66. [ Links ]
7. Barbieri RL, Makris A, Randall RW, Daniels G, Kistner RW, Ryan KJ. Insulin stimulates androgen accumulation in incubations of ovarian stroma obtained from women with hyperandrogenism. J Clin Endocrinol Metab 1986; 62: 904-10. [ Links ]
8. Harris MI, Hadden WC, Knowler WC, Bennett PH. Prevalence of diabetes and impaired glucose tolerance and plasma glucose levels in U.S. population age 20-74 yr. Diabetes 1987; 36: 523-34. [ Links ]
9. Dunaif A, Graf M, Mandeli J, Laumas V, Dobrjansky A. Characterization of groups of hyperandrogenic women with acanthosis nigricans, impaired glucose tolerance and/or hyperinsulinemia. J Clin Endocrinol Metab 1987; 65: 499-507. [ Links ]
10. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse ob gene and its human homologue. Nature 1994; 372: 425-32. [ Links ]
11. Caro JF, Sinha MK, Kolaczynski JW, Zhang PL, Considine RV. Leptin: the tale of an obesity gene. Diabetes 1996; 45: 1455-62. [ Links ]
12. Maffei M, Halaas J, Ravussin E, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight - reduced subjects. Nat Med 1995; 1: 1155-61. [ Links ]
13. Barash IA, Cheung CC, Weigle DS, et al. Leptin is a metabolic signal to the reproductive system. Endocrinology 1996; 137: 3144-7. [ Links ]
14. Segal KR, Landt M, Klein S. Relationship between insulin sensitivity and plasma leptin concentration in lean and obese men. Diabetes 1996; 45: 988-91. [ Links ]
15. Donahue RP, Prineas RJ, Donahue RD, et al. Is fasting leptin associated with insulin resistance among nondiabetic individuals? The Miami Community Health Study. Diabetes Care 1999; 22: 1092-6. [ Links ]
16. Vidal H, Auboeuf D, De Vos P, et al. The expression of ob gene is not acutely regulated by insulin and fasting in human abdominal subcutaneous adipose tissue. J Clin Invest 1995; 98: 251-5. [ Links ]
17. Dagogo-Jack S, Fanelli C, Paramore D, Brothers J, Landt M. Plasma leptin and insulin relationships in obese and nonobese humans. Diabetes 1996; 45: 695-8. [ Links ]
18. Kolaczynski JW, Goldstein BJ, Considine RV. Dexame-thasone, OB gene, and leptin in humans; effect of exoge-nous hyperinsulinemia. J Clin Endocrinol Metab 1997; 82: 3895-7. [ Links ]
19. Fernandez-Real JM, Casamitjana R, Ricart-Engel W. Leptin is involved in gender-related differences in insulin sensitivity. Clin Endocrinol (Oxf) 1998; 49: 505-11. [ Links ]
20. Jacobs HS, Conway GS. Leptin, polycystic ovaries and polycystic ovary syndrome. Hum Reprod Update 1999; 5: 166-71. [ Links ]
21. Brzechffa IA, Jakimiuk AJ, Agarwal SK et al. Serum immunoreactive leptin concentrations in women with polycystic ovary syndrome. J Clin Endocrinol Metab 1996; 81: 4166-9. [ Links ]
22. El Orabi H, Ghalia AA, Khalifa A, Mahfouz H, El Shalkani A, Shoieb N. Serum leptin as an additional possible pathogenic factor in polycystic ovary syndrome. Clin Biochem 1999; 32: 71-5. [ Links ]
23. Laughlin GA, Morales AJ, Yen SS. Serum leptin levels in women with polycystic ovary syndrome: the role of insulin resistance/hyperinsulinemia. J Clin Endocrinol Metab 1997; 82: 1692-6. [ Links ]
24. Mantzoros CHS, Dunaif A, Flier JS. Leptin concentrations in the polycystic ovary syndrome. J Clin Endocrinol Metab 1997; 82: 1687-91. [ Links ]
25. Rouru J, Anttila L, Koskinen P, et al. Serum leptin concen-trations in women with polycystic ovary syndrome. J Clin Endocrinol Metab 1997; 82: 1697-700. [ Links ]
26. Morin-Papunen LC, Koivunen RM, Tomás C, et al. Decreased serum leptin concentrations during metformin therapy in obese women with polycystic ovary syndrome. J Clin Endocrinol Metab 1998; 83: 2566-8. [ Links ]
27. Krassas GE, Kaltsas TT, Pontikides N, et al. Leptin levels in women with polycystic ovary syndrome before and after treatment with diazoxide. Eur J Endocrinol 1998; 139: 184-9. [ Links ]
28. Fleming R, Hopkinson ZE, Wallace AM, Greer IA, Sattar N. Ovarian Function and Metabolic Factors in Women with Oligomenorrhea Treated with Metformin in a Randomized Double Placebo-Controlled Trial. J Clin Endocrinol Metab 2001; 86: 1126-33. [ Links ]
29. Wabitsch M, Hauner H, Heinze E, et al. Body fat distribution and steroid hormone concentrations in obese adolescente girls before and after weigh reduction. J Clin Endocrinol Metab 1995; 80: 3469-75. [ Links ]
30. Haffner S.M., Kennedy E., Gonzalez C., Stern M.P., Miettinen H. A prospective analysis of the HOMA model. The Mexico City diabetes study. Diabetes Care 1996, 19: 1138-41. [ Links ]
31. Matsuda M., DeFronzo R.A. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 1999, 22: 1462-70. [ Links ]
32. Tai MM. A mathematical model for the determination of total area under glucose tolerance and other metabolic curves. Diabetes Care 1994; 17: 152-4. [ Links ]
33. Gower BA, Nagy TR, Goran MI, Smith A, Kent E. Leptin in Postmenopausal Women: Infuence of Hormone Therapy, Insulin, and Fat Distribution. J Clin Endocrinol Metab 2000; 85: 1770-75. [ Links ]
34. Urbanek M, Legro RS, Driscoll DA, et al. Thirty-seven candidate genes for polycystic ovary syndrome: strongest evidence for linkage is with follistatin. Proc Natl Acad Sci U S A. 1999; 96: 8573-8. [ Links ]
35. Tucci S, Futterweit W, Concepcion ES, et al. Evidence for association of polycystic ovary syndrome in caucasian women with a marker at insulin receptor gene locus. J Clin Endocrinol Metab. 2001; 86: 446-9. [ Links ]
36. Steppan CM, Bailey ST, Bhath S, et al. The hormone resistin links obesity to diabetes. Nature 2001; 409: 307-12. [ Links ]
37. Steppan CM, Lazar MA. Resistin and obesity-associated insulin resistance. Trends Endocrinol. Metab 2002; 13: 18-23. [ Links ]
38. Urbanek M, Du Y, Silander K, et al. Variation in resistin gene promoter not associated with polycystic ovary syndrome. Diabetes 2003; 52: 214-19. [ Links ]
39. Saladin R, De Vos P, Guerre-Millo M, et al. Transient increase in obese gene expression after food intake or insulin administration. Nature 1995; 377: 527-9. [ Links ]
40. Cusin I, Sainsbury A, Doyle P, Rohner-Jeanrenauld F, Jeanrenauld B. The ob gene and insulin. A relationship leading to clues to the understanding of obesity. Diabetes 1995; 44: 1467-70 [ Links ]
41. Vidal H, Auboeuf D, De Vos P, et al. The expression of ob gene is not acutely regulated by insulin and fasting in human abdominal subcutaneous adipose tissue. J Clin Invest 1996; 98: 251-5. [ Links ]
42. Kolaczynski JW, Nyce MR, Considine RV, et al. Acute and chronic effect of insulin on leptin production in humans. Diabetes 1996; 45: 699-701. [ Links ]
43. Hardie LJ, Rayner DV, Holmes S, Trayhurn P. Circulating leptin levels are modulated by fasting, cold exposure and insulin administration in lean but not Zucker (fa/fa) rats as measured by ELISA. Biochem Biophys Res Commun 1996; 223: 660-5. [ Links ]
44. Nolan JJ, Olefsky JM, Nyce MR, Considine RV, Caro JF. Effect of troglitazone on leptin production. Studies in vitro and human subjects. Diabetes 1996; 45: 1276-8. [ Links ]
45. Chehab F, Lim M, Lu R. Correction of the sterility defect in homozigous obese female mice by treatment with human recombinant leptin. Nat Genet 1996; 12: 318. [ Links ]
46. Ahima R, Dushay J, Flier SN, Prabakaran D, Flier JS. Leptin accelerates the timing of puberty in normal female mice. J Clin Invest 1997; 99: 391-5. [ Links ]
47. Mantzoros CS, Flier JS, Rogol AD. A longitudinal assess-ment of hormonal and physical alterations during normal puberty in boys. Rising leptin levels may signal the onset of puberty. J Clin Endocrinol Metab 1997; 82: 1066-70. [ Links ]
48. Rosenbaum M, Leibel L. Role of gonadal steroids in the sexual dimorphisms in body composition and circulating concentrations of leptin. J Clin Endocrinol Metab 1999; 84: 1784-9. [ Links ]
49. Quinton ND, Smith RF, Clayton PE, et al. Leptin binding activity changes with age. The link between leptin and puberty. J Clin Endocrinol Metab 1999; 84: 2336-42. [ Links ]
50. Sinha MK, Ohanessian JP, Heinman ML, et al. Nocturnal rise of leptin in lean, obese, and non-insulin dependent diabetes mellitus subjects. J Clin Invest 1996; 97: 1344-7. [ Links ]
Recibido: 26 de diciembre de 2002
Aceptado: 8 de agosto de 2003

