










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680On-line version ISSN 1669-9106
Medicina (B. Aires) vol.64 no.1 Buenos Aires Jan./Feb. 2004
La ventaja de los heterocigotas
Desde el centro de Asia, los seres humanos invadieron Europa hace unos 35.000 años. Llevaban herramientas de hueso y metal, necesarias para la caza y el arte, ventajas que contribuyeron a desplazar a los hombres de Neandertal, los primitivos residentes del continente1. Sabemos también que portaban un gen letal: el de la fibrosis quística, tan resistente y estable que se diseminó por toda Europa. ¿Cómo una enfermedad que lleva a la muerte temprana, persiste por tanto tiempo?
Las características individuales se heredan con genes que proceden de ambos progenitores. Los que codifican esas características pueden tener dos formas o alelos, una de cada uno. Si los dos alelos son iguales el individuo es homocigota, si uno es diferente, heterocigota. En los individuos homocigotas con ambos alelos defectuosos o mutados del gen de una enfermedad genética recesiva, la enfermedad se manifiesta en forma clínica. Esto no ocurre en los heterocigotas con sólo un alelo defectuoso, que en ciertas circunstancias, tienen una superioridad reproductiva sobre los homocigotas con ambos alelos normales. Es la ventaja de los heterocigotas. Ventaja ambigua, bendición o maldición, depende del ambiente donde vivan y del azar de los cruzamientos. Lo que ocurre en la malaria, la fiebre tifoidea y el cólera ayuda a entender la ventaja.
La malaria es endémica en Africa, cada treinta segundos mata una persona, mientras que el cólera y otras diarreas son las principales responsables de muerte en niños menores de cinco años. La anemia de células falciformes (drepanocítica), común en los negros, es la primera enfermedad de la cual conocimos su falla molecular. La malaria y la anemia drepanocítica comparten el glóbulo rojo como escenario de su acción. La fibrosis quística es la enfermedad genética letal más común en los caucásicos. En el cólera y otras diarreas se secreta gran cantidad de fluidos y sales por canales iónicos que son defectuosos en la fibrosis quística. Sobre esta relación tratará nuestra nota.
La anemia de células falciformes es una enfermedad hemolítica de transmisión genética, con una incidencia de cuatro por mil. La sustitución de un glutamato por valina en la cadena b de la globina es responsable de su poca solubilidad que, al precipitar, resulta en un glóbulo rojo con forma de medialuna. El gen para la cadena a está en el cromosoma 16; en el 11 el de las cadenas b y g. Los heterocigotas para la anemia tienen iguales proporciones de hemoglobina normal y anormal, no muestran signos o síntomas de la enfermedad a menos que viajen a regiones de gran altitud, donde la concentración de O2 es baja. Hasta hace poco, los individuos homocigotas para la anemia falciforme no vivían lo suficiente para dejar descendencia. Cada vez que dos alelos mutados se reunían en un homocigota, se eliminaban al no reproducirse y morir quien los tenía. En algunas poblaciones africanas, el 45% es heterocigota para esta anemia, a pesar de que la pérdida de los alelos correspondientes en los homocigotas y el reemplazo de estos alelos por nuevas mutaciones requeriría una tasa de mutación muy superior a las conocidas.
¿Cuál es la razón para que un gen dañino para el homocigota sea tan común? La respuesta fue sugerida en 1949. Al finalizar una disertación de J.B.S. Haldane en un congreso sobre Enfermedad y Evolución, G. Montalenti, genetista italiano, señaló que con L Bianco y E Silvestroni habían notado la estrecha distribución geográfica entre la talasemia y la malaria y la posible relación entre ambas2. Haldane estuvo de acuerdo con Montalenti y ese mismo año publicó un trabajo con su hipótesis de la malaria; allí no menciona a Montalenti 3. Sugirió que la mayor resistencia de los glóbulos rojos microcíticos –como los de la talasemia en soluciones hipotónicas– podría asemejarse a la ruptura de eritrocitos que el protozoario causa en la malaria.
La talasemia es una anemia hereditaria que ocurre por mutaciones que afectan la síntesis de globina a o b4. La malaria es una enfermedad con fiebre y escalofríos recurrentes relacionados con la hemólisis sincrónica de eritrocitos parasitados por organismos del género Plasmodium y cuyos vectores son los mosquitos del género Anopheles. La propagación de estos mosquitos al sur del Sahara habría ocurrido hace unos 6.000 años, siguiendo el desarrollo de la agricultura y el aumento en la población; entonces habrían aparecido los mecanismos de defensa para la enfermedad5.
Hasta aquí todo era hipótesis fundada en la distribución geográfica y la relación temporal de las enfermedades. En 1954 A.C. Allison publicó un trabajo efectuado en Africa, en una zona endémica de malaria6. Experimentó con los nativos. Tomó a 30, 15 que eran heterocigotas para la anemia de células falciformes y 15 sin el gen anómalo, y les inoculó el parásito o los hizo picar con mosquitos infectados. La parasitemia fue positiva en 2 portadores del gen anómalo y en 14 de los nativos sin ese gen. La hipótesis quedó demostrada, los heterocigotas eran más resistentes a la malaria. El eritrocito deformado es un pobre huésped para el parásito y la mayor rapidez en la destrucción de los eritrocitos con hemoglobina anormal y un menor crecimiento del parásito podrían ser las causas de esta protección7. También se ha demostrado la mayor resistencia a la malaria de las talasemias y otras formas anómalas de hemoglobina7, 8. Dos alelos para la hemoglobina anormal se eliminan en el estado homocigota y la selección positiva para el heterocigota mantiene el alelo en el reservorio génico. Por razones que se desconocen, las mujeres heterocigotas son más fértiles que las homocigotas normales.
¿Qué ventaja aporta el gen de la fibrosis quística a los heterocigotas? La enfermedad se debe a mutaciones en el gen que codifica el CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), proteína que forma un canal iónico asociado al movimiento del Cl- y ATP a través de las membranas celulares y regulador de la actividad de otros canales iónicos9. Como con otros casos, el gen y su producto se identifican con el nombre de la enfermedad que provoca su mutación o ausencia, nomenclatura confusa, impuesta y protestada.
Más de 1400 mutaciones se han descripto en el gen del CFTR, ubicado en el cromosoma 7; la eliminación de una fenilalanina –una cruel simplicidad– en la posición 508 (dF508) es la mutación más frecuente y corresponde al 70% de los casos9 (Fig. 1).
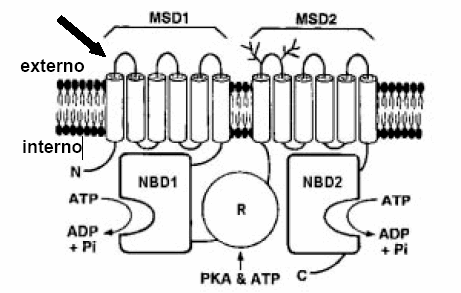
Fig. 1.- Estructura del CFTR (Cystic Fibrosis Transmembrane Conductance Regulator). La molécula tiene dos dominios que consisten cada uno en 6 segmentos transmembranales (MSD1 y MSD2) conectados por un dominio R al que se unen la proteina quinasa A (PKA) y el ATP como reguladores. Las regiones NBD1 y NBD2 permiten la unión de nucleótidos cíclicos y la hidrólisis del ATP está relacionada al funcionamiento del canal. La mutación DF508 se ubica en el dominio NBD1. La salmonella por su matriz polisacárida externa, se une al primer dominio extracelular del CFTR (flecha).
La fibrosis quística se manifiesta cuando ambos alelos están mutados, enfermedad recesiva letal que afecta a una de cada 2500 personas. El 5% de la población es heterocigota, tiene un solo alelo del CFTR mutado y es asintomática. Recesivo es una palabra de origen latino que significa apartado. Es así, el alelo permanece oculto en portadores y emerge cuando dos de éstos comienzan una familia; uno de cada cuatro hijos será un enfermo de fibrosis quística, dos serán heterocigotas y uno sano, no portador.
Los pulmones, glándulas exocrinas y diversas células como los linfocitos están comprometidas en esta enfermedad 9. La más característica alteración es la secreción mucosa que obstruye las vías aéreas y conductos de las glándulas y permite la infección, inflamación y destrucción de los tejidos. La pilocarpina, agonista colinérgico, provoca profuso sudor y es utilizada en el diagnóstico. Debido a una defectuosa absorción del ClNa, el sudor de los enfermos tiene más sal que los normales. Un adagio secular del folklore del norte de Europa dice:
Miserable es el niño que al ser besado en la frente tiene gusto a sal. Está embrujado y pronto morirá.
En 1991 se sugirió que la resistencia al cólera de los portadores heterocigotas de las mutaciones del CFTR sería responsable de la alta incidencia del alelo anormal del CFTR en las poblaciones europeas10. Por siglos, el cólera es endémico en la India; apareció en Europa en 183011. En 1817 hubo una gran epidemia en la India y avanzó hacia Europa siguiendo las corrientes humanas, ejércitos, ríos y vías comerciales terrestres y marítimas. En 1829 irrumpió en Irán; llegó a Polonia en 1830 con un ejército ruso. En 1831 los puertos de Gran Bretaña estaban contaminados. Existen descripciones de la fibrosis quística en Europa desde comienzos del siglo XVIII, antes de la llegada del cólera. El cólera, no habría seleccionado al alelo mutado del CFTR. Otros patógenos que causan diarrea lo habrían hecho, entre los candidatos, los de la urbana y endémica fiebre tifoidea.
Existen evidencias del papel del CFTR en la internalización de bacterias Gram negativas patogénas entéricas como Salmonella typhi y oportunistas como Pseudomona aeruginosa. Se ha sugerido que el alelo mutado del CFTR confiere una ventaja: los heterocigotas con una sola copia del gen mutado resisten el efecto debilitador de la fiebre y la disentería de la fiebre tifoidea12.
Las salmonellas infectan invadiendo las células epiteliales del intestino delgado, luego se trasladan a la submucosa. El traslado es parte de un proceso en el que intervienen la activación celular, la secreción de citoquinas y el proceso inflamatorio. La matriz polisacárida externa de la bacteria se une al primer dominio extracelular del CFTR y así invade el intestino (Fig. 1). La exposición de los enterocitos a la salmonella provoca, en minutos, migración de moléculas de CFTR intracelulares hacia la superficie apical, paso importante en la infección12. La copia defectuosa del CFTR en los heterocigotas impide estos procesos y de ahí la resistencia a la infección. El primer dominio extracelular del CFTR, al que se une la bacteria, no participa en las funciones de canal del CFTR, debido a que continúa activo luego de su eliminación12.
Distintas son las infecciones en las vías aéreas. En los individuos sin alteración del gen CFTR y los heterocigotas, la internalización de las bacterias facilita su eliminación por descamación de las células epiteliales, la activación de citoquinas y otras respuestas inflamatorias12. En la fibrosis quística, el mayor daño es la pérdida progresiva de la función pulmonar por infecciones crónicas debidas a P. aeruginosa. Entre el 75 y el 90% de los enfermos están infectados con esta bacteria tornada resistente a los antibióticos por cambio del fenotipo13. Con los dos alelos del CFTR mutados homocigotas, los procesos de defensa no ocurren y se acumulan las bacterias en la vía aérea.
El CFTR es una proteína inusual, no sólo forma un canal iónico, sino que es un receptor para las bacterias en las vías digestiva y aérea, un regulador de la actividad de otros canales y está presente en células epiteliales y no epiteliales14. ¿Qué consecuencias traería el bloqueo farmacológico del CFTR? En principio, un modelo experimental de fibrosis quística y también un tratamiento del cólera. En la diarrea hay una mayor secreción de Cl- por canales CFTR, dirigido hacia la luz intestinal; la toxina colérica estimula estos canales y el Na+ y el agua acompañan por la vía paracelular esta secreción aumentada de Cl- (Fig. 2). Por esto, se ensaya un inhibidor del CFTR en ratones y se demostró que bloquea en más del 90% la secreción de fluidos inducida por la toxina colérica15.

Fig. 2.- Funcionamiento del CFTR en un epitelio secretor (célula intestinal). El Cl- es incorporado a la célula por el transportador 2Cl- Na-K ubicado en la membrana basolateral. La bomba de Na-K elimina Na+ celular al medio e incorpora K+ que es reciclado por canales específicos. El Cl- se transporta hacia la luz intestinal por el CFTR y el Na+ se secreta, acompañado con H2O en la misma dirección por vía paracelular. La toxina colérica (Tox.Col) se une en forma irreversible a la adenilatociclasa, incrementa la concentración intracelular de cAMP y se activa la PKA, que regula la función del CFTR. La respuesta es la secreción de gran cantidad de Cl- , de Na+ y de H2O. El bloqueo del CFTR podría aplicarse para tratar el cólera (ver texto).
¿Qué dicen estos datos y estudios? Una paradoja, la ley de las compensaciones, se quita algo a cambio de otra cosa. El genoma es un registro escrito de nuestro pasado, una historia clínica del individuo y de las enfermedades que lo han marcado. La mutación DF508 del gen CFTR parece haberse originado en Asia y su diseminación refleja las migraciones humanas aun con cálculos teóricos supuestos y datos muy variados16. La resistencia a la fiebre tifoidea podría haber seleccionado a los heterocigotas de la mutación DF508 del CFTR y la incidencia de esta enfermedad a lo largo del tiempo puede explicar por qué tantos europeos tienen esa mutación en el CFTR.
Las bacterias de la fiebre tifoidea y el cólera necesitan de la copia normal del CFTR para introducirse en las células e infectar. La de los heterocigotas no le sirve y así, eliminan a los que tienen versiones normales del gen, seleccionan la copia distinta y conservan una versión rara y nociva de un gen.
Todo tiene su precio. Si un niño sufre de fibrosis quística o de anemia falciforme, su padre y su madre son portadores del gen anormal aunque no muestran signos de la enfermedad, son heterocigotas. En la genética de poblaciones, el individuo es sólo un reservorio temporal de material genético, el dueño de una alícuota del mismo. Así, la pérdida de los hijos homocigotas se compensa en términos demográficos con los beneficios de los padres en regiones con malaria o fiebre tifoidea. Hay que entender esto para no horrorizarse al considerar esta ventaja como una bendición.
Basilio A. Kotsias
Instituto de Investigaciones Médicas Alfredo Lanari, Facultad de Medicina, Universidad de Buenos Aires
e-mail: kotsias@mail.retina.ar
1. Wells S. The journey of man. A genetic odyssey. Princeton: Princeton University Press, 2002.
2. Haldane JBS. Disease and evolution. Ric Sci 1949; 19 (Suppl) 325-34.
3. Haldane JBS. The rate of mutation of human genes. Proc. 8th Int. Congr. Genet. Hereditas 1949; 35 (Suppl) 267-73.
4. Feliú Torres A, Bonduel M, Sciuccati G, et al. b talasemia mayor en la Argentina. Medicina (Buenos Aires) 2002; 62: 1124-34.
5. Currat M, Trabuchet G, Rees D, Perrin P, Harding RM, Clegg JB, Langaney A, Excoffier L. Molecular analysis of the b-globin gene cluster in the Niokholo Mandenka population reveals a recent origin of the bS Senegal mutation Am J Hum Genet 2002; 70: 207-23.
6. Allison AC. Protection afforded by sickle-cell traiy against subtertian malarial infection. Br Med J 1954; 1: 290-4.
7. Smith TG, Ayi K, Serghides L, Mcallister CD, Kain KC. Innate immunity to malaria caused by Plasmodium falciparum. Clin Invest Med 2002; 25: 262-72.
8. Modiano D, Luoni G, Sodiomon Sirima B, et al. Haemo-globin C protects against clinical Plasmodium falciparum malaria. Nature 2001; 414: 305-8.
9. Kunzelmann K. CFTR: interacting with everything? News Physiol Sci 2001; 16: 167-70.
10. Rodman DM, Zamudio S. The cystic fibrosis heterozygote -advantage in surviving cholera? Med Hypothesis 1991; 36: 253-8 (citado en Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 1994; 266: 107-9).
11. Kiple KF. Plague, pox and pestilence. New York: Barnes and Noble books, 1997.
12. Lyczak JB, Pier GB. Salmonella enterica Serovar Typhi modulates cell surface expression of its receptor, the cystic fibrosis transmembrane conductance regulator, on the intestinal epithelium. Infection Immunity 2002; 70: 6416–23.
13. Drenkard E, Ausubel FM. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 2002; 416: 740-3.
14. Assef YA, Damiano AE, Zotta E, Ibarra C, Kotsias BA. CFTR in K562 human leukemic cells. Am J Physiol (Cell Physiol) 2003; 285: C480-8.
15. Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJV, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest 2002; 110: 1651-8.
16. Dawson KP, Frossard PM. A hypothesis regarding the origin and spread of the cystic fibrosis mutation DF508. Q J Med 2000; 93: 313-5.

