










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.64 n.2 Ciudad Autónoma de Buenos Aires mar./abr. 2004
Distintas formas de presentación clínica de un raquitismo hipofosfatémico autosómico dominante por mutación del factor de crecimiento fibroblástico 23 en una familia
Armando Luis Negri1, Teresa Negrotti2, Guillermo Alonso3, Titania Pasqualini3
1Instituto de Investigaciones Metabólicas, Universidad del Salvador,
2Sección de Genética y
3Sección de Endocrinología, Crecimiento y Desarrollo, Departamento de Pediatría, Hospital Italiano de Buenos Aires
Dirección postal: Dr. Armando Luis Negri, Instituto de Investigaciones Metabólicas, Libertad 836, 1012 Buenos Aires, Argentina
Fax: (54-11) 5031-9703 e-mail: negri@casasco.com.ar
Resumen
Describimos distintas formas de presentación clínica de un raquitismo hipofosfatémico autosómico dominante en 4 miembros de una misma familia y su respuesta al tratamiento. Paciente N° 1: de sexo femenino de 60 años que consultó por dolores costales y pélvicos, con osteoporosis densitométrica, hipofosfatemia con bajo umbral renal de fósforo, PTH intacta normal y calcemia normal. Tratada con fósforo neutro y calcitriol logró la normalización bioquímica y una notable mejoría de la densitometría en menos de un año. Paciente N° 2: su nieta, consultó al año y ocho meses de edad por presentar talla en percentil 3 y genu varum. En el laboratorio mostró hipofosfatemia y fosfatasa alcalina total muy elevada y en la Rx de mano, ensanchamiento y deflecamiento epifisario compatible con raquitismo. Tratada con fósforo neutro y calcitriol, normalizó los parámetros bioquímicos y logró un ascenso en el percentil de talla de 3 a 50 a los 20 meses de tratamiento. Paciente N° 3: la madre de la paciente N° 2, quien sin ninguna manifestación clínica y con densitometría ósea normal presentó hipofosfatemia que se normalizó con tratamiento con fosfato neutro. Paciente N° 4: el tío de la paciente N° 2, tuvo raquitismo hipofosfatémico de niño, y luego de los 5 años normalizó el fósforo sin tratamiento. Estudiado a los 29 años presentó fósforo normal y densitometría ósea normal. El análisis del ADN genómico de la paciente N° 3 mostró una mutación con sentido erróneo en el gen del factor de crecimiento fifroblástico 23 (sustitución de arginina por una glutamina en posición 179). Por lo tanto se llegó al diagnóstico de raquitismo/osteomalacia hipofosfatémico autosómico dominante.
Palabras clave: Raquitismo hipofosfatémico autosómico dominante; Factor de crecimiento fibroblástico 23; Pérdida renal de fosfato; Raquitismos hipofosfatémicos familiares
Abstract
Different forms of clinical presentation of an autosomal dominant hypophosphatemic rickets caused by a FGF 23 mutation in one family. In this report we describe different forms of clinical presentation of an autosomal dominant hypophosphatemic rickets (ADHR) in 4 members of the same family as well as the treatment used in these patients and their response to it. Patient N° 1: a 60 year old female who consulted for bone pain. Bone densitometry showed osteoporosis. Laboratory assays showed hypophosphatemia with low renal phosphate threshold, high total alkaline phosphatase, normal intact PTH and normal serum calcium. With neutral phosphate and calcitriol, the biochemical parameters normalized and bone densitometry improved significantly in less than a year. Patient N° 2: her grand daughter consulted at 1 year and 8 months of age for growth retardation (height at percentile 3) and genu varum. Laboratory assays showed low serum phosphate and high total alkaline phosphatase; thickening and irregular epiphyseal borders of the wrists were observed radiologically. She began treatment with calcitriol and phosphorus with normalization of laboratory parameters and increase in growth (height increasing to percentile 50 after 20 months of therapy). Patient N° 3: mother of patient N° 2, she had no clinical manifestations and normal densitometry but presented low serum phosphate (1.9 mg/dl) that normalized with neutral phosphate therapy. Patient N° 4: he was the youngest son of Patient N° 1, who had had hypophosphatemic rickets, by age 5; his serum phosphate normalized without treatment. At age 29, he presented normal serum phosphate and bone densitometry. Genomic DNA analysis performed in patient N° 3, showed missense mutation with substitution of arginine at position 179 for glutamine. The family was catalogued as having autosomal dominant hypophosphatemic rickets/osteomalacia.
Key words: Autosomal dominant hypophosphatemic rickets; Fibroblast growth factor 23; Renal phosphate wasting; Hereditary hypophosphatemic rickets
La pérdida urinaria aislada de fosfato puede deberse a varios trastornos hereditarios caracterizados por un defecto en el transporte renal de fosfato que produce fosfaturia, hipofosfatemia, enfermedad ósea raquítica u osteomalácica, dolor óseo, retardo de crecimiento y abscesos dentarios. Entre estos trastornos hereditarios se encuentran el raquitismo hipofosfatémico ligado al cromosoma X (sigla en inglés XLH)1 que es el más frecuente, el raquitismo hipofosfatémico autosómico dominante (sigla en inglés ADHR)2, 3, 4, 5 y finalmente el raquitismo hipofosfatémico hereditario con hipercalciuria (sigla en inglés HHRH)6.
Aquí describimos las distintas formas de presentación clínica de una hipofosfatemia hereditaria debida a pérdida urinaria aislada de fósforo en cuatro miembros de una misma familia, en uno de los cuales se detectó una mutación con sentido erróneo en el factor de crecimiento fibroblástico 23 (FGF23) por lo que el trastorno hereditario fue catalogado como un ADHR. Describimos también el tratamiento instituido a los pacientes y su respuesta al mismo.
Casos clínicos
Paciente N° 1
Paciente del sexo femenino de 60 años de edad que consultó al Instituto de Investigaciones Metabólicas por dolores costales y pélvicos de por lo menos un año de evolución. En estudios previos mostraba osteoporosis densitométrica e hipofosfatemia. De niña había presentado genu varum y había sido tratada con radiación ultravioleta. Menopausia natural a los 50 años. Sus exámenes de laboratorio mostraban fosfatemia baja (1.7 mg/dl), fosfatasa alcalina total elevada (FAL: 863 UI/L; N<300 UI/L), PTH intacta normal y umbral renal de fósforo muy bajo (2.01 mg/dl). En la radiología de columna presentó osteopenia marcada sin fracturas. Se hizo el diagnóstico presuntivo de osteomalacia hipofosfatémica por pérdida urinaria de fosfato. Comenzó tratamiento con fósforo neutro 1.5 g por día y calcitriol 0.5 µcg/día, lográndose normalización de los parámetros bioquímicos y una notable mejoría de la densitometría: T score de L2-L4 aumentó de –3.2 a –0.5 en 9 meses; T score de cuello femoral aumentó de –3.5 a –2.1 en 19 meses.
Paciente N° 2 (nieta de la paciente N° 1)
Consultó por primera vez a la Sección de Endocrinología, Crecimiento y Desarrollo del Departamento de Pediatría del Hospital Italiano de Buenos Aires al año y 8 meses de edad por presentar talla en percentil 3. Al examen físico presentaba genu varum y leve ensanchamiento de muñecas. Los exámenes complementarios mostraron hipofosfatemia (3.0 mg/dl), gran elevación de la FAL (1755 UI/L; N < 640) PTH intacta y 1,25(OH)2 D3 normales. La Rx de mano mostró edad ósea de 18 meses con ensanchamiento y deflecamiento epifisario compatible con raquitismo. En el interrogatorio de los antecedentes familiares surgió que la abuela estaba siendo tratada con sales de fósforo y el tío había tenido un raquitismo de niño. Se hizo entonces el diagnóstico de raquitismo hipofosfatémico familiar.
Comenzó tratamiento con calcitriol y luego con sales neutras de fósforo. En sucesivos controles periódicos se advirtió crecimiento compensador, progresiva mejoría del genu varum y normalización de la fosfatemia y de los niveles de fosfatasa alcalina. A los 5 meses de tratamiento mostraba velocidad de crecimiento en percentil 97 con una ganancia de talla (percentil 10). A los 3 años y 6 meses, luego de 20 meses de tratamiento ininterrumpido alcanzó el percentil 50 de talla.
Paciente N° 3 (hija mayor del caso N° 1 y madre de la Paciente N° 2)
A raíz de los antecedentes de su hija y su madre, la paciente se estudió en el Instituto de Investigaciones Metabólicas. Al momento de la consulta tenía 33 años de edad y se encontraba asintomática. En los análisis de laboratorio presentaba hipofosfatemia (1.9 mg/dl), PTH intacta normal y 1,25 (OH)2 D3 baja, la densitometría ósea era normal. Inició tratamiento con fósforo neutro, con lo que normalizó la fosfatemia, pero luego lo abandonó. En la última consulta se presentó con un nuevo embarazo.
El 15/1/03 se realizó análisis del ADN genómico (GeneDx, Inc., Gaithersburg, MD, USA). Los exones 1-22 del gen PHEX fueron amplificados por PCR y analizados por análisis de secuenciación bidireccional. No se hallaron mutaciones cuando se comparó con la secuencia normal publicada del gen PHEX. Un polimorfismo heterocigoto fue identificado en el intrón 9 que demostró la presencia de ambos alelos en la muestra estudiada. Luego se le estudio el gen del factor de crecimiento fibroblástico 23 (FGF23). Se obtuvo y analizó la secuencia completa de los tres exones y de las regiones linderas entre exones e intrones del gen. Se observó un cambio heterocigoto de guanina a adenina en el exón 3 (CGG a CAG), lo que predice una sustitución de la arginina normal por una glutamina en la posición 179 (R179Q o Arg179Gln). Este hallazgo fue confirmado por amplificación con PCR y secuenciación del exón 3 en una segunda alícuota del ADN.
Paciente N° 4 (hijo menor de la paciente N° 1, y tío de la paciente N°2)
Al año de edad se le había diagnosticado raquitismo hipofosfatémico; no caminó hasta los 2 años de edad. Recibió tratamiento con vitamina D en altas dosis y a los 3 años y 9 meses le indicaron solución de fósforo. Siguió con la solución de fósforo en forma intermitente hasta que la abandonó. Sin medicación normalizó los valores de fósforo a los 5 años de edad. A los 29 años volvió a estudiarse a raíz de la enfermedad de su madre y sobrina. En el momento de la evaluación se encontraba asintomático, con una talla de 1.67 m; presentaba fosfatemia normal y traía una densitometría ósea normal (Z score de L2-L4: –0.8).
Discusión
En este informe clínico presentamos una hipofosfatemia hereditaria por pérdida urinaria de fosfato producida por mutación del FGF 23 que tuvo distintas formas de presentación clínica en los miembros de la familia portadora (Fig. 1). La paciente N° 1 llegó a la consulta con reducción importante de la masa ósea, dolores óseos e hipofosfatemia. Al presentar un umbral renal de fósforo muy bajo se hizo el diagnóstico de hipofosfatemia por pérdida urinaria de fosfato. La notable mejoría de la masa ósea especialmente en columna lumbar inducida por el tratamiento con fosfato neutro y calcitriol en el período menor de un año, hizo pensar que se trataba de una osteomalacia y no de una osteoporosis postmenopáusica.
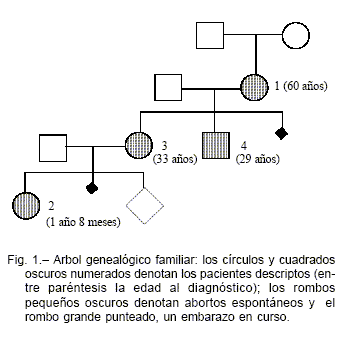
En su nieta, paciente N° 2, al consultar por retardo de crecimiento, se constataron signos clínicos (genu varum), radiológicos y de laboratorio de raquitismo hipofosfatémico a los 20 meses de edad. Con el tratamiento de fósforo neutro y calcitriol, mejoró su crecimiento y sus alteraciones óseas. En ese momento se decidió estudiar a la madre de la niña, paciente N° 3 e hija mayor de la paciente N°1, quien estaba asintomática y no había tenido trastornos óseos durante la infancia, detectándose una masa ósea normal para la edad asociada a presencia de hipofosfatemia. Finalmente el paciente N° 4, hijo menor de la paciente N° 1 se encontraba asintomático, con masa ósea normal y con fósforo sérico normal; sin embargo de niño había presentado raquitismo hipofosfatémico.
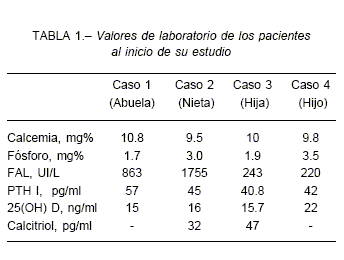
Los valores séricos de fosfato se encontraron disminuidos en 3 de los 4 pacientes (Tabla 1). Los valores de calcio y hormona paratiroidea fueron normales en todos nuestros pacientes, como se describe clásicamente en el raquitismo hipofosfatémico y que lo diferencia del raquitismo por deficiencia de vitamina D, del raquitismo dependiente y del resistente a vitamina D. Los valores de calcitriol sérico fueron normales en presencia de hipofosfatemia en dos de los cuatro miembros de la familia; esto es un elemento característico del raquitismo ligado al X y del autosómico dominante, debido a un trastorno en la regulación de la 1-alfa hidroxilasa renal.
Como señalamos en la introducción, existen distintos tipos de raquitismo/osteomalacia hipofosfatémico familiar. El raquitismo hipofosfatémico ligado al cromosoma X (sigla en inglés XLH)1, es la forma más común y se hereda en forma dominante1-7. Se ha demostrado recientemente que mutaciones inactivadoras en el gen que regula el fósforo con homología de las endopeptidasas, localizado en el cromosoma Xp22.1 (sigla en inglés PHEX) es responsable del XLH8. El raquitismo hipofosfatémico autosómico dominante (sigla en inglés ADHR)4, 5, 6 es una forma menos común de raquitismo hipofosfatémico que presenta disturbios bioquímicos comparables al XLH, pero es autosómico dominante y tiene penetrancia variable4, 5, 6. Recientemente se ha encontrado que estas familias presentan mutaciones con sentido erróneo en el factor de crecimiento fibroblástico 23 (FGF23)9, localizado en el cromosoma 12p13.3. Se ha propuesto que las mutaciones afectan los sitios de clivaje necesarios para una normal degradación de FGF23, prolongando por lo tanto su actividad biológica10, 11 .
El FGF23 recombinante induce hipofosfatemia debida a pérdida urinaria de fosfato in vivo11, 12, 13. Además, la inyección de FGF23 en ratones suprime la expresión de la 1-alfa hidroxilasa de la 25 (OH) vitamina D. Esto explicaría por qué los niveles de 1,25 (OH)2D3 no están aumentados en los casos de aumento de FGF23 a pesar de la hipofosfatemia que es un gran estímulo de la 1-alfa hidroxilación de la 25 (OH) vitamina D. Los tumores productores de osteomalacia oncogénica y células que expresan el gen salvaje de FGF23 producen al menos dos formas moleculares de FGF23, una de tamaño completo que es una proteína de 32 kD y un fragmento de 12 kD. Dado que solamente el fragmento más grande fue hallado en algunas mutaciones productoras de raquitismo hipofosfatémico autosómico dominante, se ha propuesto que las mutaciones afectan los sitios de clivaje necesarios para una normal degradación de FGF23, prolongando por lo tanto su actividad biológica11, 12, 14. Se ha propuesto que el FGF23 es un sustrato para el PHEX, que se halla mutado en el raquitismo XLH8. Por lo tanto, distintos mecanismos moleculares como: a) generación de un FGF23 mutado resistente a su degradación o b) alteración de la degradación del FGF23 por reducción o pérdida de la actividad del PHEX o c) la sobreproducción de FGF23 por tumores responsables de la osteomalacia oncogénica, pueden producir elevación de la concentración de FGF23 y consecuentemente pérdida urinaria de fósforo.
A pesar que existe gran variabilidad en la manifestación fenotípica de todos los raquitismos hipofosfatémicos, debido a que la normalización del fósforo sérico con el tratamiento con fósforo oral y la mejoría del trastorno de mineralización ósea son menos usuales en la forma clásica de XLH15, 16, es que nos pareció probable que esta familia tuviera la forma autosómica dominante, ADHR. Los pacientes que padecen este trastorno se agrupan en dos formas clínicas: un grupo que presenta pérdida renal aislada de fósforo, dolor óseo, debilidad muscular y a veces fracturas pero sin deformidades de las extremidades inferiores como adolescentes y adultos. El otro grupo se presenta en la primera infancia con pérdida de fósforo, raquitismo, trastorno del crecimiento y deformidades de las piernas. Mientras que en la mayor parte de los pacientes la hipofosfatemia persiste en la edad adulta, algunos pierden el trastorno de reabsorción de fosfato luego de la pubertad, como parece haber ocurrido en el paciente N° 4.
En enero de 2003 el ADN genómico de la paciente N° 2 fue estudiado (GeneDx, Inc., Gaithersburg, MD, USA) para determinar si era la posible causa de su hipofosfatemia. Debido a que la frecuencia de mutaciones es mayor en el gen PHEX, y a pesar que por la clínica y la respuesta al tratamiento se había pensado que podía tratarse de una alteración del gen del FGF23, se analizó en primer lugar los exones 1-22 del gen PHEX por secuenciación bidireccional, no hallándose mutaciones. Este análisis detecta más de ¾ de las mutaciones en los pacientes con XLH familiares y 1/3 en los casos con mutaciones esporádicas17, 18. Se encaró entonces el análisis del gen que codifica el FGF23, que detectó una mutación R179Q en el gen FGF23 que causa el reemplazo de la arginina normal por una glutamina en la proteína resultante. Esta mutación con sentido erróneo ha sido informada previamente en este raquitismo hipofosfatémico10. La presencia de esta mutación confirmó el diagnóstico de raquitismo hipofosfatémico autosómico dominante. El tratamiento combinado con dosis adecuadas de fosfato oral, 1-3 gramos por día (20-60 mg/kg/día) en dosis divididas, y de calcitriol 0.25-4 µg/día (30-65 ng/kg/día) logró la curación del raquitismo y la osteomalacia, con mejoría en el crecimiento y desaparición de los dolores óseos.
1. Winters RW, Graham JB, Williams TF, et al.: A genetic study of familial hypophosphatemia and vitamin D-resistant rickets with review of the literature. Medicine 1958; 37: 97-142. [ Links ]
2. Bianchine JW, Stambler AA, Harrison HE: Familial hypophosphatemic rickets showing autosomal dominant inheritance. Birth Defects Or Artic Ser 1971; 7: 287-94. [ Links ]
3. David L, Pesso JL, Cochat P, Plauchu H, Francois R: Rachitisme hypophosphatémique autosomique type Scriver: Une observation familiale. Pédiatrie 1987; 42: 563-8. [ Links ]
4. Econs MJ, McEnery PT: Autosomal dominant hypophosphatemic rickets/osteomalacia: Clinical characterization of a novel renal phosphate wasting disorder. J Clin Endocrinol Metab 1997; 8: 674-81. [ Links ]
5. Scriver CR, MacDonald W, Reade T, Glorieux FH, Nogrady B: Hypophosphatemic nonrachitic bone disease: An entity distinct from X-linked hypophophatemia in the renal defect, bone involvement, and inheritance. Am J Med Genet 1977; 1: 101-17. [ Links ]
6. Tieder M, Modai D, Samuel R, et al.: Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med 1985; 312: 611-6. [ Links ]
7. Scriver CR, Reade TM, DeLuca HF, Hamstra AJ: Serum 1,25-dihydroxyvitamin D levels in normal subjects and in patients with hereditary rickets or bone disease. N Engl J Med 1978; 299: 976-9. [ Links ]
8. HYP Consortium: A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 1995; 11: 130-6. [ Links ]
9. ADHR Consortium: Autosomal dominant hypophosphatemic rickets is associated with mutations in FGF23. Nat Genet 2000; 26: 345-8. [ Links ]
10. White KE, Carn G, Lorenz-Depiereux B, Benet-pages A, Strom TM, Econs MJ: Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF 23. Kidney Int 2001; 60: 2079-86. [ Links ]
11. Bai XY, Miao D, Goltzman D, Karaplis AC: The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic clevage and enhances biological potency. J Biol Chem 2003; 278: 9843-9. [ Links ]
12. Shimada T, Mizutani S, Muto T y col. Cloning and characterization of FGF 23 as a causative factor for tumor induced ostemalacia. Proc Natl Acad of Sci USA 2001; 98: 6500-5. [ Links ]
13. Shimada T, Yoneya T, Hino R, Takeuchi Y, Fukumoto S, Yamashita T: Transgenic mice overexpressing fibroblast growth factor 23 (FGF 23) demostrate hypophosphatemia with low serum 1,25-dihydroxyvitamin D [1,25(OH)2D] and rickets/osteomalacia. J Bone Miner Res 2001; 16 Suppl: S151 abstract. [ Links ]
14. Shimada T, MutoT, UrakawaI et al.: Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic clevage and causes hypophosphatemia in vivo. Endocrinology 2002; 143: 3179-82. [ Links ]
15. Kruse K, Hinkel GK, Griefhan B: Calcium metabolism and growth during early treatment of children with X-linked hypophosphatemic rickets. Eur J Pediatr 1998; 157: 894-900. [ Links ]
16. Friedman NE, Lobaugh B, Drezner MK: Effects of calcitriol and phosphorus therapy on the growth of patients with X-linked hypophosphatemia. J Clin Endocrinol Metab 1993; 76: 839-44. [ Links ]
17. Holm IA, Nelson AE, Robinsin BG, Mason RS, Marsh DJ, Cowell CT, Carpenter TO. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab 2001; 86: 3889-99. [ Links ]
18. Christie PT, Harding B, Nesbit MA, Whyte MP, Thakker RV. X-linked hypophosphatemia attributable to pseudoexons of the PHEX gene. J Clin Endocrinol Metab 2001; 86: 3840-4. [ Links ]
Recibido: 15-08-2003
Aceptado: 23-10-2003

