










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.64 n.3 Buenos Aires mayo/jun. 2004
Mutations in CFTR gene and clinical correlation in Argentine patients with congenital bilateral absence of the vas deferens
Estrella M. Levy1, Patricia Granados1, Vanesa Rawe 2, Santiago Brugo Olmedo2, María C.Luna1, Eduardo Cafferata1, Omar H. Pivetta1
1 Departamento de Genética Experimental, Centro Nacional de Genética Médica, ANLIS, Carlos G. Malbrán;
2CEGYR, Centro de Estudios en Ginecología y Reproducción, Buenos Aires
Postal address: Dra. María Cecilia Luna, Departamento de Genética Experimental, Centro Nacional de Genética Médica, Las Heras 2670, 1425 Buenos Aires, Argentina. Fax: 54-11-4801-4428. e-mail: cl@fibertel.com.ar
Abstract
Congenital bilateral absence of the vas deferens (CBAVD) is a form of male infertility in which mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene have been identified. Here we identify different mutations of CFTR and the poly-T variant of intron 8 (IVS8) in Argentine patients and analyze sweat test values and clinical characteristic related to Cystic Fibrosis (CF). For counseling purposes the two most frequent mutations in Argentine CF population: DF508 and G542X were screened in wives. In all cases, it was possible to reduce the risk of CF/CBAVD descendants in these couples because none of the mutation were found in the 36 samples. Eight patients (23%) showed abnormal chloride values (> 60 mmol/l). A second group of 6 patients (18%) had borderline values of sweat chloride (40-59 mmol/l). We defined another group with 6 patients (18%), with normal sweat chloride levels (30-39 mmo/l) and a fourth group of 14 (41%) patients with sweat chloride below 30 mmol/l. DF508, the most frequent CF mutation in the Argentine population, was found on 15 of the 72 chromosomes (21%), R117H mutation was detected on 2 of 62 chromosomes (3%). Only one R347P allele was found on 28 chromosomes analyzed (2%). On a sample of 27 patients, IVS8 analysis showed a frequency of 6/56 chromosomes (11%) of 5T allele. Even though these findings present an improvement in the detection of mutations related to clinical correlations in Argentine CBAVD population, the search for other common and uncommon mutations should be continued.
Key words: Cystic fibrosis; CBAVD; CFTR; Vas deferens; Male infertility
Resumen
Correlación de las características clínicas con mutaciones del gen CFTR en pacientes argentinos con ausencia bilateral congénita de vasos deferentes. La ausencia bilateral congénita de vasos deferentes (CBAVD) es una forma de infertilidad masculina en la que se han identificado mutaciones en el gen de la conductancia transmembrana de la fibrosis quística (CFTR). Hemos estudiado en pacientes argentinos diferentes mutaciones en el CFTR y la variante poli T del intron 8 (IVS8) y analizado los valores de test del sudor y las características clínicas relacionadas a la Fibrosis Quística (FQ). Para el asesoramiento genético se han estudiado en las esposas de estos pacientes, las dos mutaciones más frecuentes en la población FQ del país, DF508 y G542X. Como no se encontraron mutaciones, el riesgo de descendencia CF/CBAVD fue reducido del 2 al 0.7%. Ocho pacientes (23%) presentaban test del sudor anormales (> 60 mmol/l). Un segundo grupo de 6 pacientes (18%) presentaron valores dudosos (40-59 mmol/l). Hemos definido un tercer grupo de 6 pacientes con valores normales de test del sudor (18%), comprendidos entre los 30 y 39 mmo/l, y un cuarto grupo de 14 pacientes (41%) con valores de cloruro en sudor inferiores a 30 mmol/l. La mutación más frecuente en la población CF argentina, DF508, fue encontrada en 15 de los 72 cromosomas (21%) analizados, la R117H fue encontrada en 2 de los 62 cromosomas estudiados (3%). Un único alelo R347P fue encontrado en los 28 cromosomas analizados (2%). De los 27 pacientes a los que se les estudió el tracto IVS8, 6/56 cromosomas (11%) presentaban el alelo 5T. Si bien estos hallazgos representan un avance en relación a la detección de mutaciones correlacionadas con los síntomas clínicos en la población CBAVD argentina, se debe continuar la búsqueda de otras mutaciones comunes y raras con el fin de establecer una conducta terapéutica en estos pacientes.
Palabras clave: Fibrosis quística; CBAVD; CFTR; Vasos deferentes; Infertilidad masculina
Congenital bilateral absence of the vas deferens (CBAVD) occurs in 1-2% of men with infertility1. Obstruction of the Wolffian ducts results in the absence or atrophy of the vas deferens, epididymal body and tail, seminal vesicles and ejaculatory ducts2. CBAVD is an obstructive azoospermia, inherited in an autosomal recessive way3,4. Although isolated CBAVD is considered a distinct clinical entity5; it was suggested in 1971 that some males with CBAVD might have a mild form of Cystic Fibrosis (CF)6. This hypothesis was corroborated after the identification and cloning of the cystic fibrosis transmembrane conductance regulator (CFTR) gene7. The observation that mutations in the CFTR gene are present in men with CBAVD has led to the proposal that CBAVD may be a primary genital form of CF8, 9. The classic form of CF is a disorder characterized by chronic pulmonary disease, pancreatic exocrine insufficiency, elevated concentrations of electrolytes in sweat, infertility in males (95% incidence) because of obstructive azoospermia10.
Different reports have shown a high frequency of CFTR mutations in CBAVD patients11-14. The 5T allele in intron 8 of the CFTR gene leads to a higher proportion of mRNA transcripts lacking exon 9 than the two others alleles, 7T and 9T. The 5T variant is a mutation frequently associated to the CBAVD phenotype15. However, many studied CBAVD patients did not seem to carry mutations in both copies of the gene. This may be explained by the high molecular heterogeneity of CBAVD and the different spectrum of CFTR mutations when compared with CF patients16.
At present, for Latin American populations, there is not enough data on the molecular basis of CBAVD. The ethnic origin of the Argentine population is heterogeneous and geographically unevenly distributed17. Currently, no information is available about the real frequency and distribution of mutations in the CFTR gene of CBAVD Argentine patients. For these reason, the purpose of this study was to investigate the prevalence of 12 of the most frequent CF mutations and the IVS8 polymorphism in Argentine CBAVD patients, and the most distinctive CF clinical characteristics, including the sweat test and possible correlations with their genotype.
Moreover, patients with CBAVD are infertile because of obstructive azoospermia. Assisted reproduction techniques allows these patients to inseminate18. Men with CBAVD, however, have an increased risk of CF if their partners also carry a CF mutation. Therefore to render more accurate counseling, we screened the two most frequent mutations in Argentine CF population: DF50819 and G542X20 in wives.
Materials and Methods
Patients
Our study group comprised 36 unrelated infertile men (35 CBAVD and 1 CUAVD, congenital unilateral absence of vas deferens) undergoing fertility treatment (since July, 1997 until April 2001) who were diagnosed as having obstructive azoospermia. All patients were found to have absence of the vas deferens by scrotal exploration and clinical observation of impalpable vasa. Absence of seminal vesicles was confirmed in most cases by biochemical semen analysis (lower concentration of fructose, low volume, and low pH). The patients were 26-50 years of age (average 34.6) and were born in Argentina. None of them had a familial history of cystic fibrosis.
We also included the 36 wives of the patients, who also had no familial history of cystic fibrosis.
Sweat tests
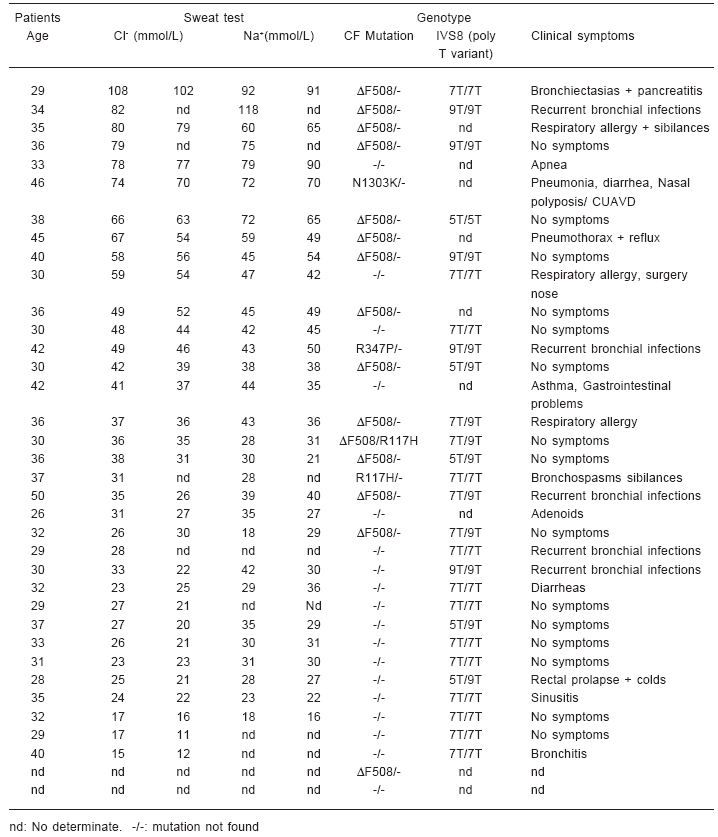
Sweat test were performed using iontoforesis with pylocarpine stimulation21. In most of the patients it was possible to determine the chloride and sodium ion concentrations (Table 1).
Table 1.– Summary of the clinical and genotype findings in a population of 34 CBAVD and one CUAVD patients
Clinical picture
Respiratory and gastrointestinal symptoms as well as a familial history of CF, were reported using a model questionnaire.
Mutation analysis
Genomic DNA was extracted from peripheral blood leukocytes by standard procedures22. The DF508 (exon 10), G542X (exon 11) and N1303K (exon 21) mutations were detected by allele specifics - polymerase chain reaction AS-PCR method23.
To identify other cystic fibrosis mutations in this patients, amplification of exons 4, 7, 11, 19 and 20 and the flanking sequences were performed by PCR as previously described by Zielinski24. After amplification, R334W, R347P (exon 7), G551D, R553X (exon 11), and R1162X (exon 19) mutations were identified by restriction enzyme analysis as previously described by Gasparini25, 26 and Shrimpton27.
The intronic mutation 3849 +10Kb C®T was analyzed as described by Highsmith28.
R117H (exon 4), 1717-1G®A (intron 10) and W1282X (exon 20) were identified by hybridization with allele specific oligonucleotides22. The probes sequences were obtained from Shrimpton27 and Kerem29.
The technique of detection of the CFTR intron 8 polypirimidine tract length variants was modified from Chillon15.
Statistical analysis
Differences between frequencies were tested by the c2 statistic with Epi-info program for personal computers (version 6).
Results
The results of clinical and genetic data of the 36 patients (35 CBAVD and one CUAVD) are summarized in Table 1, where the patients have been listed in decreasing order of average of two independents sweat chloride levels.
Sweat test and clinical picture
Sweat test was performed on 34 of the 36 patients, with a mean Cl- value of 42.4 mmol/l (range 13.5-105 mmol/l) and a mean Na+ value of 46 mmol/l; (range 17-118 mmol/l), Na+ was measured in 30 patients. The sweat test is considered positive for CF when the chloride concentrations is higher than 60 mmol/l10. Borderline values are between 40-59 mmol/l. Although values lower than 39 mmol/l are considered normal. Our results allow us to discriminate among four groups with respect to sweat chloride levels.
The sweat test was abnormal in 8 of 34 patients (23%). Seven of these, presented at least one mutation (88%). One patient was 5T/5T homozygous. The patient with the higher sweat test value (108-102 mmol/l) had chronic pancreatitis and bronchiectasis. The others 5 patients, who presented clinic symptoms of CF, presented pulmonary symptoms such as pneumothorax and recurrent bronchial infections. The CUAVD patient was included in this group because he had chloride value of 72 mmol/l (74-70 mmol/l).
A second group of 6 patients (18%) had borderline values of sweat chloride. Four of these patients had at least one mutation. Only one patient presented the 5T allele of the IVS8 polymorphism.
We defined another group with 6 patients (18%), with normal sweat chloride levels (30-39mmo/l) Five of them had at least one CF mutation. In this group it was possible to perform the complete genotyping of one patient, DF508/R117H, who did not have any CF symptoms. Four of the 6 patients in this group reported respiratory problems.
The last group of 14 (41%) patients had a sweat chloride below 30 mmol/l, and only one patient with 28 mmol/l (30-26 mmol/l) had a CFTR DF508 mutation.
Two patients could not, or refused to, undergo the whole protocol.
Recurrent respiratory problems only, including bronchitis, sinusitis, nasal polyps, etc, were detected in 13/34 patients (38%). Gastrointestinal disorders only, in general recurrent diarrheas, were referred by 1/34 patients (3%). Both, respiratory and gastrointestinal symptoms were referred by 5/34 patients (15%).
Biochemical analysis and urogenital abnormalities
All patients were found to have absence of the vas deferens by scrotal exploration and the clinical observation of impalpable vasa. The patient with CUAVD, had the other vas disrupted by traumatic inflamation.
Azoospermia was present in all patients. Semen analysis showed all volumes < 1 ml, pH < 7.0, fructose < 8 mmol/l and citrate < 10 mmol/l, in accordance with previous reports11.
Genetic analysis
Of the 36 patients with CBAVD screened for mutations in the CFTR gene, 20 (55%) had at least one CFTR mutation.
The overall frequency of CF mutations detected DF508, which was found on 15 of 20 patients with a detectable mutation (75%); DF508 is also the most frequent CF mutation in the Argentine CF population19. The other mutations found were: R117H in 2 of 31 patients screened, one combined with DF508 and R347P in one patient. The presence of the 5T polymorphism was found in 6 of the 27 patients with CBAVD (22%), one of them homozygous, 5T/5T. The patient with CUAVD had the N1303K mutation (Table 1).
For counseling purposes the two most frequent mutations in Argentine CF population: DF508 and G542X were investigated in the CBAVD patients wives, but no mutation was found in any of the 36 samples.
Discussion
In the present study we describe the genotypic and clinical phenotypic characteristics of an Argentine CBAVD population. More than half of the patients with CBAVD (54%) and the patient with CUAVD had at least one detected CFTR mutation.
Previous studies screening for CFTR gene mutations in CBAVD patients have reported different frequency of patients having one or two mutations11-14. However our results completely agree with Attardo30 in Sicilian population. Using the same panel of mutations, we identified two mutations in 57.6% of the patients with CF coming from the same geographical area, a single mutation in 34.4% and no mutation in the remaining 8%. DF508 is the most frequent CF mutation worldwide31 and in Argentina (64%)19 and is also the most frequent CFTR mutation in our CBAVD patients (21%), followed by R117H present in two of the 58 chromosomes screened (3.5%). R117H has been found with elevated frequency in these patients32, 33. This mutation has not been reported previously in patients with CF in Argentina. R117H and the R347P were detected in this study for first time in Argentine CF or CBAVD populations.
A single nucleotide substitutions may have a profound effect on the splicing efficiency inducing both exon inclusion and skipping. The changes in the splicing pattern were modulated by the composition of the polimorphic TG/T locus in intron 834. The variant 5T, an important pathogenetic factor for CBAVD, was present in five of the 27 patients analyzed (18.5%), one of wich was homozygous.
All patients with CBAVD came for evaluation of infertility, and no other symptoms were referred spontaneously. However, an accurate evaluation of the history of the patient revealed that many patients with CBAVD had suffered from or were at the time complaining of disturbances similar to those commonly encountered in patients with CF. In our sample seven patients (21%) and the CUAVD patient had an abnormal sweat test and six CBAVD (18%) had borderline values. Respiratory problems like nasal polyps and recurrent bronchial infections were reported by 38% of the patients, digestive alterations were described by 3%, and both respiratory and digestives simultaneously by 15% of CBAVD. Eighty percent of patients with sweat test over 30 mmol/l had at least one CF mutation. Only two patient DF508/- were not included in this group, one with a chloride value of 28 mmol/l (30-28 mmol/l), no sweat test was available for the other.
A proportion of 10-30% of CBAVD males with no detectable CFTR mutation have been noted by others11, 12, 15, 35. Argentina has an ethnic mixture with a Mediterranean profile. Accordingly mutations that appear principally in Spain and Italy should receive special emphasis in genetic analysis. However, a significant contribution of native CF alleles should be considered in the characterization of CFTR gene in Argentine CBAVD patients.
Genetic counseling remains difficult in couples with CBAVD infertility due to the wide spectrum of mutations. When we screened for the two most frequent mutations in the Argentine CF population in the patients wives, it was in all cases to reduce the apparent risk of descendans CF/CBAVD in these couples. Based in the Bayes theorem, as in our area the prevalence of CF carriers is about 0.04, since our routine panel in women covers 67% of CF mutations, we predict a risk a priori of a CF offspring equal to 1 x 0.04 x 0.5 = 0.02, or a combined risk of 1 x 1 x 0.5 = 0.5 when the female partner is positive and 1 x 0.0135 x 0.5 = 0.007 when she is negative for DF508 or G542X mutations.
These data provide a better characterization of CBAVD patients in Argentina; however, the search for other common and uncommon mutations should be continued.
Acknowledgements: We thank Dr. Paul Quinton for the critical review of the manuscript and his scientific support and advice; Dr. Friedman KJ for DNA controls to the IVS8 alleles, Dr. Carolina Haefliger and Dr. Viviana Varela for their technical support. This work was supported by a grant from Asociación Fipan and Fundación Alberto J. Roemmers, Buenos Aires, Argentina.
1. Jequier AM, Ansell ID, Bullimore NJ. Congenital absence of the vasa deferentia presenting with infertility. J Androl 1985; 6: 15-9. [ Links ]
2. Taussig LM, Lobeck CC, Di SaintÁgnese PA, et al. Fertility in males with cystic fibrosis. N Engl J Med 1972; 287: 586-9. [ Links ]
3. Nelson RE Congenital absence of the vas deferens: a review of the literature and report of three cases. J Urol 1950; 63: 176-82. [ Links ]
4. Schellen TMCM, Stratten A van. Autosomal recessive hereditary congenital aplasia of the vasa deferentia in four siblings. Fertil Steril 1980; 35: 401-4. [ Links ]
5. McKusick VA: OMIM TM Mendelian Inheritance in Man TM. http://www.ncbi.nih.gov 08/01/04. [ Links ]
6. Holsclaw DS, Perlmutter AD, Jeckin H, et al. Genital abnormalities in male patients with cystic fibrosis. J Urol 1971; 106: 568-74. [ Links ]
7. Kerem B, Roemmens JM, Buchanan JA, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245: 1073-80. [ Links ]
8. Dumur V, Gervais R, Rigot JM, et al. Abnormal distribution of CF DF508 allele in azoospermic men with congenital aplasia of epididymis and vas deferens. Lancet 1990; 336: 512. [ Links ]
9. Anguiano A, Oates RD, Amos JA, et al. Congenital bilateral absence of vas deferens: a primarily genital form of cystic fibrosis. JAMA 1992; 267: 1794-7. [ Links ]
10. Boat TJ, Webb MJ, and Beaudet AL Cystic fibrosis. In: Scriver, CR, Beaudet AL, Sly WJ and Valle D (eds), Metabolic Basis of Inherited Diseases. 6th Ed. New York: McGraw 1989 p 2649-80. [ Links ]
11. Casals T, Bassas LI, Ruiz-Romero J, et al. Extensive analisys of 40 infertile patients with congenital absence of vas deferens: in 50% of cases only one CFTR allele could be detected. Hum Genet 1995; 95: 205-80. [ Links ]
12. Costes B, Girodon E, Ghanem N, et al. Frequent ocurrence of the CFTR intron 8 (TG)n 5T allele in men with congenital bilateral absence of vas deferens. Eur J Hum Gene: 1995; 3: 285-93. [ Links ]
13. Rave-Harel N, Madgar I, et al. CFTR haplotype analysis reveals generic heterogeneity in the etiology of congenital bilateral aplasia of the vas deferens. Am J Hum Genet 1995; 56: 1359-66. [ Links ]
14. Dörk T, Dworniczak B, Aulehla-Scholz C, et al. Distinct spectrum of CFTR gene mutations in congenital absence of vas deferens. Hum Genet 1997; 100: 365-77. [ Links ]
15. Chillón M, Casals T, Mercier B, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med 1995; 332: 1475-80. [ Links ]
16. Casals T, Bassas L, Egozcue S, et al. Heterogeneity for mutations in the CFTR gene and clinical correlations in patients with congenital absence of the vas deferens. Hum Reprod 2000; 7: 1476-83. [ Links ]
17. Palatnik M. Seroantropología argentina. Sangre 1966; 11: 395-412. [ Links ]
18. Silber SJ, Ord T, Balmaceda J, et al. Congenital absence of vas deferens. The fertilizing capacity of human epididymal sperm. N Engl J Med 1990: 323: 1788-92. [ Links ]
19. Luna MC, Granados PA, Olek K, et al. Cystic fibrosis in Argentina: the frequency of the DF508 mutation. Hum Genet 1996; 97: 314. [ Links ]
20. Chertkoff L, Visich A, Bienvenu T, et al. Spectrum of CFTR mutations in Argentine cystic fibrosis patients. Clin Genet 1997; 51: 43-7. [ Links ]
21. Gibson LE, Cooke RE. A test for concentration of electro-lytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine iontophoresis. Pediatrics 1959; 23: 545-9. [ Links ]
22. Sambrook J, Fritsch EF, Maniatis T. Isolation of DNA from Mammalian Cells (for blood samples). In Molecular Cloning (a laboratory manual), 2nd ed. Cold Spring Harbor Lab Press 1989; 9.16-9.21. [ Links ]
23. Fortina P, Conant R, Monokian G, et al. Non-radioactive detection of the most common mutations in the cystic fibrosis transmembrane conductance regulator gene by multiplex allele-specific polymerase chain reaction. Hum Genet 1992; 90: 375-8. [ Links ]
24. Zielinski J, Rozmahel R, Bozon D, et al. Genomic DNA sequence of the cystic fibrosis transmembrane conduc-tance regulator (CFTR) gene. Genomics 1991; 10: 214-28. [ Links ]
25. Gasparini P, Nunes V, Savoia A, et al. The search for South European cystic fibrosis mutations: identification of two new mutationss, four variants and intronic sequences. Genomics 1991; 10: 193-200. [ Links ]
26. Gasparini P, Marigo C, Bisceglia G, et al. Screening of 62 mutations in a cohort of cystic fibrosis patients from north eastern Italy: their incidence and clinical features of defined genotypes. Hum Mutat 1993; 2: 389-94. [ Links ]
27. Shrimpton AE, McIntosh I, Brock DJH. The incidence of the different cystic fibrosis mutations in the Scottish population: effects on prenatal diagnosis and genetic counselling. J Med Genet 1991; 28: 317-22. [ Links ]
28. Higshmith WE, Burch LH, Zhou Z, et al. A novel mutation in the cystic fibrosis gene in patients with pulmonary disease but normal sweat chloride concentrations. N Engl J. Med 1994; 331: 974-80. [ Links ]
29. Kerem B, Zielinski J, Markiewicz D et al. Identification of mutations in regions corresponding to the two putative (ATP) binding folds of the cystic fibrosis gene. Proc Natl Acad Sci USA 1990; 87: 8447-51. [ Links ]
30. Attardo T, Vicari E, Mollica F, et al. Genetic, andrological and clinical characteristics of patients with congenital bilateral absence of the vas deferens. Int J Androl 2001; 24: 73-9. [ Links ]
31. Cystic Fibrosis Genetic Analysis Consortium. http:/ www.genet.sickkids.on.ca 10/11/03. [ Links ]
32. Gervais R, Dumur V, Rigot JM, et al. High frequency of the R117H cystic fibrosis mutation in patients with congenital absence of the vas deferens. N Engl J Med 1993; 328: 446-7. [ Links ]
33. Patrizio P, Asch RH, Handelin B, et al. Aetiology of congenital absence of vas deferens: genetic study of three generations. Hum Reprod 1993; 8: 215-20. [ Links ]
34. Pagani F, Buratti E, Stuani C, et al. Missense, nonsense and neutral mutations define juxtaposed regulatory elements of splicing in CFTR exon 9. JBC Papers in press. Published on May 5, 2003 as Manuscript M212813200 [ Links ]
35. Zielinski J, Patrizio P, Corey M, et al. CFTR gene variant for patients with congenital absence of the vas deferens. Am J Hum Genet 1995; 57: 958-60. [ Links ]
Received: 20-08-2003
Accepted: 14-01-2004


{kind=link}