










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.66 n.2 Buenos Aires mar./abr. 2006
Periventricular nodular and subcortical neuronal heterotopia in adult epileptic patients
Damián E. Consalvo1, 2, Silvia S. Kochen1, Agustín Alurralde1, Estela Centurion1, Pablo A. Salgado1, Patricia Saidon1, Roberto E. P. Sica1
1Centro de Referencia de Epilepsia, División Neurología, Hospital Ramos Mejía; 2Fundación FEMIEN (Fundación para el Estudio de las Enfermedades de la Mielina y Neurooncológicas)
Dirección postal: Dr. Damián E. Consalvo, Centro de Referencia de Epilepsia, División de Neurología, Hospital J. M. Ramos Mejía, Urquiza 609, 1221 Buenos Aires, Argentina. Fax: (54-11) 4932-6101. E-mail: dconsalvo@fibertel.com.ar
Abstract
Developmental malformations are brain abnormalities that occur during embryogenesis. Neuronal migration disorders, including heterotopic lesions, constitute one type of such abnormalities. The aim of the study was to compare the epileptic clinical patterns of patients with periventricular nodular heterotopia (PNH) (G1) with those affected by subcortical heterotopia (SCH) (G2) looking for differences between both groups which, eventually, might suggest the type of the underlying malformation. The variables studied in both groups were: type of the heterotopia depicted on MRI studies, sex, age, age at seizure onset, annual seizure frequency, localization of the ictal symptomatogenic zone, characteristics of the EEG, other associated anomalies on the magnetic resonance images (MRI) besides the heterotopia, and response to treatment. The only difference found between both groups was the type of heterotopia as shown by MRI studies. The other assessed variables did not significantly (p>0.05) differ between groups. No differences in the clinical features characterizing epilepsy could be found in patients with PNH or SCH, being the images the only tool able to differentiate them.
Key words: developmental malformations, MRI, periventricular nodular heterotopia, subcortical nodular heterotopia
Resumen
Heterotopía neuronal nodular y subcortical en pacientes adultos con epilepsia. Las malformaciones de la corteza cerebral son un grupo de entidades que se producen durante las etapas del desarrollo embrionario y cuya manifestación clínica puede ser la epilepsia. Estas malformaciones pueden ser diagnosticadas in vivo a través de las imágenes por resonancia magnética (IRM). Un subtipo particular de éstas lo constituyen los trastornos en la migración neuronal, dentro de los cuales se ubican las heterotopías (HT). El objetivo del estudio fue comparar enfermos portadores de HT periventriculares (G1) con aquellos portadores de HT subcorticales (G2). Se analizaron las variables sexo, edad y edad de inicio de la epilepsia (EI) en años, antecedentes familiares (AF) o prenatales (AP), frecuencia anual de crisis (FAC) y características semiológicas de las crisis, hallazgos en el EEG e IRM y respuesta al tratamiento farmacológico. G1 (n=13): 8 mujeres (61.5%), edad promedio 32.9 ± 11.5 (rango 20-59), EI 13.7 ± 7.6 (rango 2-23), AF 1 caso (7.7%), AP en 1 (7.7%), FAC 28.3 ± 31.4 (rango 0-120), crisis multifocales en 5 (38.5%), crisis temporales en 5 pacientes (38.5%), EEG epileptiforme (EEGE) en 7 casos (53.8%), anomalías asociadas en las IRM (AAIRM) en 8 sujetos (61.5%) y 4 casos refractarios al tratamiento (30.7%). G2 (n=8): 6 mujeres (75%), edad promedio 30 ± 9.7 (rango 13-43), EI 11.1 ± 6.3 (rango 1-19), AP 2 (25%), FAC 30 ± 39.5 (rango 0-120), crisis multifocales en 4 sujetos (50%), crisis temporales en 5 pacientes (62.5%), EEGE en 7 casos (87.5%), AAIRM en 3 casos (37.5%) y 1 caso refractario al tratamiento (12.5%). El análisis de las diferentes variables clínicas analizadas no mostró diferencias significativas entre ambos grupos, siendo las imágenes el único elemento que permitió su diferenciación.
Palabras clave: malformaciones del desarrollo, resonancia magnética, heterotopías nodulares periventriculares, heterotopías nodulares subcorticales
Malformations of cerebral cortical development encompass a heterogeneous group of disorders frequently recognized on magnetic resonance images (MRI). These types of disorders are a cause of human epilepsy1-4.
According to the period of neurogenesis where developmental disruptions occur, they are classified into three groups2, 3: abnormal neuronal and/or glial proliferation lacking apoptosis, abnormal neuronal migration and abnormal cortical organization.
Dysfunctions affecting the mechanisms of cortical proliferation, migration or organization lead to different MRI findings. Lissencephalies, subcortical band heterotopia spectrum, cobblestone complex and heterotopia are specifically included within the group of neuronal migration disorders3.
Neuronal migration abnormalities are due to disruption of the mechanisms allowing neurons to move from the proliferative or germinal zone, situated surrounding theventricular region, to their final location into the cerebral cortex5, 6.
The migration mechanism is complex. Different authors have attempted to explain it on the basis of the so-called "radial unit" hypothesis, according to which migration occurs following the columnar model in which the neurons located at the ventricles walls are organized5, 7, 8. These neurons follow a radial pattern direction and are guided by specialized glial cells, although some cases of tangential or non-radial migration, involving GABAergic interneurons, have been identified as well5,7-11.
Migration abnormalities yield heterotopia. Patients with heterotopia could be grouped into two different major categories, those showing subependymal nodules of gray matter located at the lateral ventricles walls, known as periventricular nodular heterotopia (PNH), and those others carrying heterotopic lesions located at subcortical regions, named subcortical heterotopia (SCH), usually extending from the ventricular surface up to the cortex3, 12, 13. Probably, both types of disorders develop at the same chronobiological period during cerebral cortical development2, 3. Another subgroup of heterotopic lesions comprises those subjects with marginal glioneuronal heterotopia; in these cases, the diagnosis can only be achieved by pathological examination; some authors have considered this last subgroup as being part of the microdysgenesis group3, 14, 15.
The ectopic gray matter results from an insult that occurred during the late neuronal proliferating period, which allows the multiplication of neuroblasts but prevents their normal migration from the paraventricular region. Arrested migration of neuroblasts, in their way to the cortex, is the cause of the subcortical nodular heterotopia13. It is not yet clear which are the factors conditioning the appearance of heterotopia, probably several events contributefor their occurrence3, 10, 16.
Recent studies have demonstrated the association between PNH and a mutation in the X-linked gene encoding filamin-1 (FLN1), which is located on chromosome Xq28. FLN1 is a protein that modulates actin organization, which is needed for cellular locomotion during migration. The presence of this mutation in the X chromosome suggests that patients with this kind of disorder might have a family history of epilepsy10, 17-20.
However, the sole presence of heterotopia not necessarily signals the development of epilepsy. Lange et al.21 recently suggested that these abnormal gray matter foci may integrate within the normal circuits of the brain, without yielding abnormal features.
Due to the fact that patients with PNH and SCH can be individualized accurately by MRIs, we decided to compare those two groups trying to identify epileptic clinical features which may signal the presence of one or other type of malformation within their brains.
Part of the material here studied, was presented elsewhere22.
Materials and Methods
A retrospective analysis was undertaken focusing on the clinical records corresponding to those epileptic patients attending the Epilepsy Center of the Neurological Division at the Ramos Mejía Hospital, for whom MRI studies were available. We selected 73 cases with the diagnosis of cortical developmental malformations according to the classification of Barkovich et al.2, 3, 21 out of them, whose diagnosis by MRIs were PNH or SCH, were enrolled in this study.
Clinical features
On the basis of an interview addressed to each patient (n: 18) or, else, answered by a family member (n: 3), the brain ictal symptomatogenic zone (ISZ) was identified23. According to this information, the location of the ISZ in the cerebral lobe presumably affected was established. Acknowledgement for this conception required in every patient that the starting signs or symptoms of the seizures were always the same along the last two years of follow-up, which was the established period of observation.
When assessing the pregnancy histories of the patients, we specifically asked for the occurrence of immediate maternal prenatal or, else, perinatal clinical abnormalities, or the appearance of any putative abnormal event along the whole period of their own pregnancies or of other pregnancies carried out by his/her mother.
During the interview a detailed family history searching for epilepsy was obtained as well.
MRI features
The MRI studies were carried out according to the Argentine Neurological Society (SNA) and the International League Against Epilepsy (ILAE) imaging protocol guidelines24, 25.
The diagnosis of PNH was defined on the basis of the identification in all the MRI sequences of nodular or laminar tissues, isointense to the gray matter, located, subependimally, at the lateral ventricles walls. Deeply analyzing this group, it became apparent that it could be further divided into two different subgroups; diffuse PNH (Fig. 1), which was defined as multiple subependymal nodules lining both lateral ventricles, and focal PNH (Fig. 2), which was recognized by the presence of singles or multiples nodules of gray matter located at one or both lateral ventricles walls.

Fig. 1.- Inversion recovery (left) and T2-weighted (right) MRI sequences in patient # 2 showing a diffuse PNH (arrows).
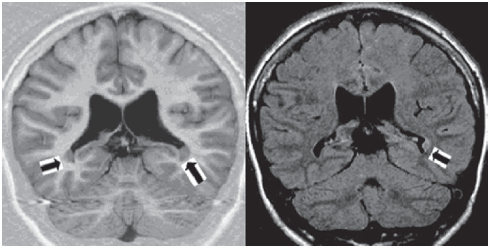
Fig. 2.- IR sequence (left) and flair (right) MRI sequences in patient # 7 showing a temporo-occipital PNH on both sides (arrows).
The diagnosis of SCH was based on the presence, in all the MRI sequences, of multinodular masses of tissue, isointense to the gray matter, subcortically located and extending from the ventricular surface to the cortex (Figs. 3 and 4).

Fig. 3.- IR sequence (left) and T2-weighted (right) MRIs sequences showing multinodular subcortical masses on the right temporo-parieto-occipital region in patient # 2 affected by SCH (arrows).

Fig. 4.- 3 D reconstruction of the brain of patient # 2 showing shallow sulci on the overlying cortex at the right hemisphere
In both circumstances, once the anatomical localization was established, a search was done looking for any other structural brain abnormality which could be related to the heterotopia. Their anatomical localization was determined and the putative associated structural abnormality defined.
EEG studies
Two or more interictal-EEG recordings were performed in each patient, following the 10-20 international recording system. Epileptic activity was defined by the presence of sharp waves, spikes or spike-and-waves complexes with a generalized or focal location.
Video-EEG was performed only in one patient belonging to the PNH group, because he was refractory to treatment; while other patient, bearing SCH, was also submitted to the same procedure for better definingher diagnosis.
Response to pharmacological treatment
Patients were considered refractory to treatment when seizure control was not achieved despite of having been medicated with one of the traditional drugs included within the first-line group of antiepileptic drugs, such as carbamazepine, phenobarbital, phenytoin and valproic acid, alone or combined with any of the more recently introduced antiepileptic drugs, such as lamo-trigine, topiramate, gabapentin, vigatrine, oxcarbazepine and levetiracetam.
Statistical Analysis
Mann-Whitney, ANOVA and c2, together with the Fishers exact test, were employed for the analysis of variables such as sex, age, age at seizure onset (AO), annual seizure frequency (ASF), seizures clinical features, EEG and MRI findings, and response to treatment.
Results
Group 1, PNH patients
This group comprised 13 subjects, 8 out of them were women (61.5%), the mean age for the whole group was 32.9 ± 11.5 years (range 20-59), while the average AO was 13.7 ± 7.6 years (range 2-23).
Only one female patient (# 2 – Table 1) within this group had had a son who was affected by febrile seizures. One other female patient (# 6 – Table 1) had had two normal pregnancies lasting 40 weeks each, but both fetuses were dead at the time of the delivery. A male patient (# 12 – Table 1) reported that his own delivery was premature, at the 37th week of his mothers pregnancy.
TABLE 1.- Clinical, electroencephalogram (EEG) and magnetic resonance images (MRI) findings in patients with PNH
The neurological examination was normal in all the subjects.
The average ASF during the period of observation, for the entire group, was 28.3 ± 31.4 (range 0-120). One patient (# 9 – Table 1), considered epileptic by hisclinical history and EEG findings, was seizure free along the whole time of the observational period.
On clinical grounds in 5 patients (38.5%) the ISZ was situated at the temporal lobe (#s 2, 3, 5, 7, 9 – Table 1), in 2 (15.3%) it was located at the occipital lobe (#s 11, 12) and in other case (7.7%) it could be localized at the parietal lobe (# 8 – Table 1). The remaining 5 subjects (38.5%) had, on clinical grounds, several foci able to arouse a seizure (#s 1, 4, 6, 10, 13 - Table 1).
Interictal EEGs showed focal epileptiform discharges in 7 cases (53.8%). In 5 of them the epileptic activity was recorded at the temporal lobe in 3out of these 5 subjects it was unilateral (#s 3, 6, 9 – Table 1), while in the other 2 it was bilateral (#s 4, 5 - Table 1). In one case the interictal discharges were recorded at the left central region (# 10 – Table1) and, in the remaining case, the epileptiform discharges were recorded at both fronto-temporal regions (# 7 – Table1). In 4 (57.1%) of the 7 patients with abnormal EEGs, the discharges were originated at ISZ clinically depicted (#s 3, 5, 9, 10 – Table 1).
Video-EEG was performed only in one patient within this group (# 1 – Table 1), the procedure allowed to recognize the multifocal origin of his discharges.
In 7 cases the placement of the nodules, as shown by MRIs, was focal and located at both lateral ventricles walls (#s 4, 5, 7, 8, 9, 10, 12 – Table 1), in 5 (#s 1, 2, 6, 11, 13 – Table 1) it was diffuse, while in the remaining patient it was focal, restricted to the frontal horn and the atrium of the left hemisphere (# 3 – Table 1).
In 8 cases (61.5%) the MRIs showed others accompanying anomalies. In 4 subjects (#s 1, 8, 10, 11 – Table 1) such abnormalities were localized at the overlying parieto-occipital cortex, which appeared thinner and with shallow sulci; hippocampal sclerosis was recognized in 2 patients, bilateral in 1 (# 1 – Table 1) and on the right side in the other (# 3 – Table 1 ), agenesis and hypoplastic corpus callosum were identified in 2 other subjects respectively (#s 8, 12 – Table 1), cerebellar hypoplasia in 1 (# 2 – Table 1), Arnold-Chiari type I malformations in 1 (# 12 – Table 1) and megacisterna magna in another (# 6 – Table 1). In three patients more than one of the citedanomalies could be found (#s 1, 8, 12 – Table 1).
Four patients (30.7%) (#s 1, 3, 4, 7 – Table 1) were refractory to drug therapy. The remaining 8 patients (#s 2, 5, 6, 8, 10, 11, 12, 13) had a good control of the seizures with the antiepileptic drugs.
Group 2, SCH patients
Eight subjects, encompassing 6 women (75%), were included within this group; the mean age for the whole groupwas 30 ± 9.7 years (range 13-43). The average AO was 11.1 ± 6.3 years (range 1-19).
Two patients had difficulties at their own deliveries; one of them (# 1 – Table 2) was born at the 36th week of gestation, while the other (# 6 – Table 2) had suffered from hypoxia at the time of her delivery, apparently due to the presence of a twin who died immediately after she was born.
TABLE 2.- Clinical, EEG and MRI findings in patients with SCH
The neurological examination was normal in all the subjects.
The average ASF was 30 ± 39.5 (range 0-120). Two patients, considered epileptic by their clinical histories and EEG findings, were seizure free along the whole period of observation.
In 5 cases (62.5%) the ISZ was localized at the temporal lobe (#s 1, 5, 6, 7, 8 – Table 2), in 2 of them it was multifocal at the same temporal lobe; in 1 case (12.5%) the ISZ was located at the parietal lobe (# 4 – Table 2) and in 1 case (12.5%) it was multifocal affecting the same frontal lobe (# 3 – Table 2). In the remaining case (12.5%) seizures arose from multiple foci, as it was suggested by the obtained clinical data (# 2 – Table 2).
In 7 cases (87.5%) the interictal EEG showed epileptic focal discharges. In 4 of them the discharges were located at the temporal lobe, in 3 were unilateral (2 on the right, #s 1.7 and 1 on the left, # 6 – Table 2) and in one bilateral (# 5 – Table 2). In one case the discharges were recorded at the right parietal lobe (# 2 – Table 2), in another at the frontal lobe, bilaterally (# 3 – Table 2), and in the remaining patient at the right temporo-parietal region (# 8 – Table 2).
In patients with abnormal EEG records, the areas where the abnormal discharges originated topographically matched the ISZ clinically detected (#s 1, 2, 3, 5, 6, 7, 8 – Table 2).
Video-EEG was performed only in one patient (# 7 – Table 2), with a doubtful diagnosis, aiming to obtain more accurate information in regards to her clinical condition; the results allowed the diagnosis of epilepsy with focal seizures onset at the temporal neocortex.
Associated abnormalities on the MRIs were found in 3 cases (37.5%). In 2 of them (#s 2, 8 – Table 2)the anomalies were situated at the overlying cortex, which appeared thinner and with shallow sulci; while in the third subject a corpus callosum agenesis was found (# 4 – Table 2).
Only one case (12.5%) was considered to be refractory to drug therapy (# 2 – Table 2). The remaining 5 (#s 1, 3, 4, 6, 8) patients had a good control of the seizures with the antiepileptic drugs.
Comparison between groups
The comparison of the studied variables between these two groups, namely sex, mean age, AO, ASF, localization of the ISZ in the temporal lobe, the presence of the multifocal seizures, epileptic abnormalities on the EEG, the presence of associated abnormalities on the MRI and the response to treatment, showed no significant differences (p ³ 0.05) for any of the comparison done.
Discussion
The findings presented here showed that it was not possible, based only on clinical and/or EEG features, recognize the presence of one or other type of heterotopia. So, when compare PNH and SCH groups no statistical differences were found amongst the different variables studied, leaving only the image as the only tool to identify the abnormalities.
However, some observations done along this study deserve some comments.
Unlike the PNH group, which seems to develop due to an abnormal genetic mechanism17-20, in the SCH group an ischemic phenomenon occurring at the vascular boundary territories, such as the region between the middle and posterior cerebral arteries, would account for the development of this last type of disorder26. This hypothesis seems to be in line with our findings, done in all the patients included within this group, because the anatomical localization of their lesions, depicted on their MRIs, coincided with areas where two artery territories overlapped. Furthermore, a past history of difficulties during pregnancy was confirmed in two of our subjects, including an earlier delivery in one case and a complicated twins pregnancy in the other, suggesting that an ischemic phenomenon might have occurred.
The presence of abnormalities in the overlying cortex, in some of our patients, suggest that this disorder occurs at the same time as the heterotopia developes or, alternatively, that an initial putative noxa would trigger off a cascade of phenomena in which the default in a previous step would lead to a deficiency in the subsequent steps and, consequently, in the final result, which is the formation of the cerebral cortex10, 27. This sort of combination between heterotopia and disruptions of the overlying cerebral cortex was seen in 30% of the patients comprising our PNH group and in 25% of those included within the SCH group.
Although the anatomical malformations are present since birth, the reason why these patients begin to suffer seizures after a long period is a question that requires further research. Other factors, perhaps environmental or genetics, in addition to the morphological anomalies, have to contribute, so that the neurons and circuits involved are given a chance to learn how to be epileptic16, 28.
The accurate identification of the ISZ has been difficult to establish for both groups of patients. In the PNH group, 38.4% of the affected people had different types of seizures, although most of them had predominant onset at the temporal lobe. Several authors have shown that seizures may start at the subependymal nodules as well as at the cortex where the neurons, clustered in those nodules, should have migrated27, 29, 30. Also the discharges may originate at a certain distance from the place where the nodules are situated, such as the hippocampus, producing a remote epileptogenic focus yielding a dual pathology13, 31. This situation illustrates the difficulty to offer this group of patients a surgical alternative for their epilepsy in the event of failure to drug treatments32.
In the SCH group, 57% of the subjects presented with different types of seizures, although the epileptogenic zones were very close one another, suggesting a clinical correlation with the malformations depicted in the imaging studies. Although the epileptogenic zones in this group were more restricted than in the subjects with PNH, they were yet enough large to require an evaluation for the possibility of surgical treatment, bearing in mind the complexity of the neuronal circuits that were probably involved33.
Most of our patients have shown an excellent response to pharmacological treatment, particularly in the SCH group. Two of the 4 patients, belonging to the PNH group, who were refractory to treatment, showed, in the images, an associated hippocampal sclerosis. The presence of dual pathology in these patients might be the reason for the difficulty in obtaining good responses to drug treatments34, 35.
Finally, and summarizing the results obtained in this study, when comparing the different clinical, EEG and MRIs findings in these two groups of epileptic patients, it was observed that the only overt difference that could be found was the picture depicted on the MRIs, which constituted the sole argument to cluster this population in two groups, the PNH and the SCH, because neither the clinical manifestations nor the EEG recordings could individualized the abnormality underlying the presence of epilepsy.
References
1. Kuzniecky R. MRI in cerebral developmental malformations and epilepsy. Magn Reson Imag 1995; 13: 1137-45. [ Links ]
2. Barkovich A, Kuzniecky R, Dobyns W, Jackson G, Becker L, Evrard P. A classification scheme for malformations of cortical development. Neuropediatrics 1996; 27: 59-63. [ Links ]
3. Barkovich A, Kuzniecky R, Jackson G, Guerrini R, Dobyns W. Classification system for malformations of cortical development: Update 2001. Neurology 2001; 57: 2168-78. [ Links ]
4. Guerrini R, Carrozzo R. Epilepsy and genetic malformations of the cerebral cortex. Am J Med Genet 2001; 106: 160-73. [ Links ]
5. Flint A, Kriegstein A. Mechanisms underlying neuronal migration disorders and epilepsy. Curr Opin Neurol 1997; 10: 92-7. [ Links ]
6. Kriegstein A. Cortical neurogenesis and its disorders. Curr Opin Neurol 1996; 9: 113-7. [ Links ]
7. Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci 1995; 18: 383-8. [ Links ]
8. Rakic P. Guidance of neurons migrating to the fetal monkey neocortex. Brain Res 1971; 33: 471-6. [ Links ]
9. Germano I, Sperber E. Transplancentally induced neuronal migration disorders: An animal model for the study of the epilepsies. J Neurosci Res 1998; 51: 473-88. [ Links ]
10. Santi M, Golden J. Periventricular heterotopia may result from radial glial fiber disruption. J Neuropathol Exp Neurol 2001; 60: 856-62. [ Links ]
11. Pilz D, Stoodley N, Golden J. Neuronal migration, cerebral cortical development, and cerebral cortical anomalies. J Neuropathol Exp Neurol 2002; 61: 1-11. [ Links ]
12. Barkovich J, Kuzniecky R. Gray matter heterotopia. Neurology 2000; 55: 1603-8. [ Links ]
13. Dubeau F, Tampieri D, Lee N, et al. Periventricular and subcortical nodular heterotopia. A study of 33 patients. Brain 1995; 118: 1273-87. [ Links ]
14. Mischel O, Nguyen L, Vinters H. Cerebral cortical dysplasia associated with pediatric epilepsy. Review on neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol 1995; 54: 137-53. [ Links ]
15. Palmini A, Lüders H. Classification issues in malformations caused by abnormalities of cortical development. Neurosurg Clin N Am 2002; 37: 1-16. [ Links ]
16. Crino P, Miyata H, Vinters H. Neurodevelopmental disorders as a cause of seizures: neuropathologic, genetic, and mechanistic considerations. Brain Pathol 2002; 12: 212-33. [ Links ]
17. Eksioglu Y, Scheffer I, Cardenas P, et al. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron 1996; 16: 77-87. [ Links ]
18. Huttenlocher P, Taravath S, Mojtahedi S. Periventricular heterotopia and epilepsy. Neurology 1994; 44: 451-5. [ Links ]
19. Guerrini R, Carrozzo R. Epileptogenic brain malformation: clinical presentation, malformative patterns and indications for genetic testing. Seizure 2001; 10: 523-43. [ Links ]
20. Sheen V, Dixon P, Fox J, et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet 2001; 10: 1775-83. [ Links ]
21. Lange M, Winner B, Müller J, et al. Functional imaging in PNH caused by a new FilaminA mutation. Neurology 2004; 62: 151-2. [ Links ]
22. Consalvo D, Alurralde A, Oddo S, et al. Heterotopías periventriculares nodulares y subcorticales: su comparación en una población de pacientes adultos con epilepsia. Medicina (Buenos Aires) 2004; 64 (Supl II): 130. [ Links ]
23. Rosenow F, Lüders H. Presurgical evaluation of epilepsy. Brain 2001; 124: 1683-700. [ Links ]
24. Grupo de neuroimágenes de la Sociedad Neurológica Argentina. Protocolo sugerido para el estudio de los pacientes con diagnóstico presuntivo de epilepsia. Rev Neurol Arg 1997; 22: 26-7. [ Links ]
25. Neuroimaging commission of the ILAE. Recommendations for neuroimaging of patients with epilepsy. Commission on neuroimaging of the International League Against Epilepsy. Epilepsia 1997; 38: 1255-6. [ Links ]
26. Battaglia G, Granata T, Farina L, DIncerti L, Franceschetti S, Avanzini G. Periventricular nodular heterotopia: epileptogenic findings. Epilepsia 1997; 38: 1173-82. [ Links ]
27. Hannan A, Servotte S, Katsnelson A, et al. Characterization of nodular neuronal heterotopia in children. Brain 1999; 122: 219-38. [ Links ]
28. Jacobs K, Kharazia V, Prince D. Mechanisms underlying epileptogenesis in cortical malformations. Epilepsy Res 1999; 36: 165-88. [ Links ]
29. Kothare S, VanLandingham K, Armon C, Luther J, Friedman A, Radtke R. Seizure onset from periventricular nodular heterotopias: depth-electrode study. Neurology 1998; 51: 1723-7. [ Links ]
30. Li L, Dubeau F, Andermann F, et al. Periventricular nodular heterotopia and intractable temporal lobe epilepsy: poor outcome after temporal lobe resection. Ann Neurol 1997; 41: 662-8. [ Links ]
31. Raymond A, Fish D, Stevens J, Cook M, Sisodiya S, Shorvon S. Association of hippocampal sclerosis with cortical dysgenesis in patients with epilepsy. Neurology 1994; 44: 1841-5. [ Links ]
32. Sisodiya S. Surgery for malformations of cortical development causing epilepsy. Brain 2000; 123: 1075-91. [ Links ]
33. Francione S, Kahane P, Tassi L, et al. Stereo-EEG of interictal and ictal electrical activity of a histologically proved heterotopic gray matter associated with partial epilepsy. Electroencephalogr Clin Neurophysiol 1994; 90: 284-90. [ Links ]
34. Li L, Cendes F, Andermann F, et al. Surgical outcome in patients with epilepsy and dual pathology. Brain 1999; 122: 799-805. [ Links ]
35. Semah F, Picot M, Adam C, et al. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998; 51: 1256-62. [ Links ]
Received: 12-07-2005
Accepted: 30-11-2005

