










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.66 n.3 Buenos Aires mayo/jun. 2006
Atontamiento miocárdico luego del accidente cerebrovascular isquémico
Daniel Varela, Fernanda Díaz, Alejandro Hlavnicka, Néstor Wainsztein, Ramón Leiguarda
Servicio de Terapia Intensiva, Fundación para la Lucha contra las Enfermedades Neurológicas de la Infancia (FLENI), Buenos Aires
Dirección postal: Dr. Néstor Wainsztein, Terapia Intensiva, Fundación para la Lucha contra las Enfermedades Neurológicas de la Infancia (FLENI), Montañeses 2325, 1428 Buenos Aires, Argentina. Fax: (54-11) 5777-3209
E-mail: nwainsztein@fleni.org.ar
Resumen
El atontamiento miocárdico, definido como una disfunción contráctil transitoria del miocardio, ha sido demostrado claramente en distintas situaciones clínicas. Sin embargo, el atontamiento miocárdico asociado a un accidente cerebrovascular isquémico ha sido escasamente notificado. Describiremos dos pacientes con diagnóstico de accidente cerebrovascular isquémico agudo que presentaron cambios electrocardiográficos, elevación enzimática y trastornos en la motilidad cardíaca, compatibles con disfunción miocárdica. Simultáneamente desarrollaron injuria pulmonar aguda rápidamente reversible, originada probablemente por un doble componente neurocardiogénico.
Palabras clave: Atontamiento miocárdico, Accidente cerebrovascular isquémico, Injuria pulmonar
Abstract
Stunned myocardium after acute ischemic stroke. The so-called stunned myocardium, defined as transitory myocardial contractile dysfunction, has been clearly demonstrated in diverse clinical situations. However, stunned myocardium related to ischemic stroke has been poorly identified. We describe two patients with diagnosis of acute ischemic stroke who developed elctrocardiographic changes, cardiac enzyme increasing levels and myocardial dysfunction secondary to abnormal cardiac wall motion. At the same time the patients developed acute lung injury with rapid resolution, perhaps as a consequence of neurocardiogenic components.
Key words: Stunned myocardium, Ischemic stroke, Lung injury
El concepto de miocardio atontado fue introducido por primera vez por Heyndrick et al en 1975, definido como miocardio viable que exhibe una prolongada disfunción ventricular izquierda, luego de la resolución de un discreto y transitorio episodio de isquemia, sin evidencias de necrosis1. Posteriormente se introduce el concepto de miocardio atontado neurogénico, definido como disfunción contráctil transitoria secundaria a una injuria neurológica aguda en pacientes con arterias coronarias normales. Esta entidad se ha descripto frecuentemente en hemorragia subaracnoidea (HSA), traumatismo de cráneo, hemorragias y tumores intracerebrales, epilepsia, meningitis, etc2. Sin embargo, el atontamiento miocárdico asociado a accidente cerebrovascular isquémico agudo ha sido escasamente notificado3.
Describiremos dos pacientes que presentaron atontamiento miocárdico secundario a accidentes cerebrovasculares isquémicos vinculados a disecciones carotídeas.
Casos clínicos
Caso I
Paciente de 44 años de edad, con antecedente de aneurismas cerebrales múltiples. Ingresó a terapia intensiva en post operatorio de cirugía endovascular con colocación de stent en la base de un aneurisma carótido oftálmico izquierdo, sufriendo disección carótidea izquierda. Al segundo día presentó paresia braquial derecha grave, la tomografía computada (TC) objetivó hipodensidad en territorio de la arteria silviana izquierda compatible con isquemia. Evolucionó con inestabilidad hemodinámica, taquicardia, taquipnea, hipoxemia grave (PaO2/FiO2: 96) requirió asistencia respiratoria mecánica (ARM). El electrocardiograma (ECG) mostró infradesnivel del segmento ST en DII, DIII, aVF, V4, V5 y V6. Presentó aumento de la CPK: 574 UI/l(VN:24-170 UI/l); MB: 48 UI/l(VN:0-25 UI/l); troponina T: 0.8 ng/ml (VN: 0.00-0.01 ng/ml). En la radiografía de tórax se observó infiltrados bilaterales difusos alveoloinstersticiales. El ecocardiograma evidenció hipoquinesia inferobasal y posterior del ventrículo izquierdo (VI), deterioro moderado de la función sistólica e insuficiencia mitral moderada. Requirió soporte inotrópico con noradrenalina y balance negativo de fluidos. Hacia el cuarto día evolucionó con mejoría progresiva de la relación PaO2/FiO2, disminución de los infiltrados en la radiografía de tórax y destete progresivo de la ARM. El día 11 normalizó el ECG y el ecocardiograma mostró VI de dimensiones, espesor y función sistólica global y segmentaria normales, sin lesiones valvulares. El día 12 de internación la paciente es derivada a otra institución, vigilante, lúcida y con una paresia braquial derecha leve.

Fig. 1.- Tomografía de cerebro del Caso I que muestra signos indirectos de isquemia en el territorio de la arteria cerebral media izquierda.
Caso II
Paciente de 30 años de edad sin antecedentes patológicos, que consultó por cefalea súbita parietal izquierda seguida de afasia de expresión, hemiparesia facio-braquio-crural derecha de 40 minutos de duración. La resonancia magnética (RM) de cerebro objetivó isquemia reciente en el territorio de la arteria silviana izquierda. El Doppler de vasos de cuello mostró hipoflujo marcado de carótida primitiva e interna izquierda, y la arteriografía de vasos de cuello disección de la arteria carótida primitiva izquierda, comenzando con tratamiento anticoagulante. Al 3er día presentó nuevo episodio de afasia y progresión del déficit motor, la RM de cerebro objetivó extensión del área isquémica. Evolucionó con disnea, taquipnea, hipoxemia grave (PaO2/FiO2:100) e infiltrados pulmonares bilaterales difusos en la radiografía de tórax, requirió ventilación no invasiva (VNI) en forma permanente durante las primeras 24 hs, apoyo inotrópico y balance negativo de fluidos. En el ECG se observó ondas T negativa e infradesnivel del segmento ST en DII, DIII, aVF, V4, V5 y V6; acompañado de elevación de enzimas cardíacas: CPK: 226 UI/l (VN: 24-170 UI/l), MB:41 UI/l (VN: 0-25 UI/l) y troponina T: 0.13 ng/ml.
El ecocardiograma mostró hipoquinesia grave del septo medial y basal, disquinesia inferobasal, con depresión leve de la función del VI. Al 5° día de evolución se retiró progresivamente la VNI y luego el soporte inotrópico. En el noveno día el ecocardiograma mostró buena función del VI sin trastornos de la motilidad global, ni segmentaria, ni dilatación de cavidades. El ECG se normalizó. La paciente fue dada de alta al 10° día de internación, persistiendo con un déficit motor braquiocrural leve y disfasia.

Fig. 2.- El electrocardiograma del Caso I permite observar infradesnivel del segmento ST en DII, DIII, aVf, V4, V5 y V6.
Discusión
El atontamiento miocárdico de origen neurogénico es un síndrome reversible que ocurre luego de injuria grave del sistema nervioso central, en ausencia de enfermedad de las arterias coronarias. Este síndrome se caracteriza por su inicio rápido luego de algún tipo de injuria neurológica, alteraciones características en el ECG, elevación de las enzimas cardiacas como CPK, CPK-MB, troponinas y alteraciones segmentarias de la pared del VI. La asociación de este síndrome con una enfermedad neurológica aguda grave, en ausencia de factores de riesgo coronario en pacientes jóvenes, cumple con los criterios para el diagnóstico de atontamiento miocárdico neurogénico.
Desde el punto de vista fisiopatológico existen múltiples estudios que demuestran que la estimulación de ciertas zonas del cerebro como la corteza insular, hipotálamo y amígdalas cerebrales producen aumento de la actividad simpática y producción excesiva de catecolaminas4, 5. Esto generaría un estímulo directo sobre la fibra miocárdica desencadenando miocitolisis y disfunción contráctil, secundario a la proteolisis selectiva de la troponina I6.
Las alteraciones electrocardiográficas secundarias a enfermedades neurológicas fueron descriptas inicialmente en 1938 y se observaron especialmente asociadas a HSA en aproximadamente el 50-100% de los casos7, 8; no obstante fueron descriptas secundarias a otros tipos de enfermedades neurológicas como lesiones intracerebrales ocupantes de espacio, convulsiones y meningitis. Alteraciones como inversión simétrica de la onda T, prolongación del segmento QT, depresión o elevación del segmento ST, son las más comúnmente observadas.
Conjuntamente con los cambios electrocardiográficos se produce elevación de enzimas cardíacas como CPK-MB y troponinas, siendo esto un signo fidedigno de injuria miocárdica. La elevación de la troponina I ha demostrado ser un marcador específico de disfunción miocárdica en HSA aneurismática, y de mortalidad en pacientes con lesión neurológica aguda9.

Fig. 3.- Resonancia Magnética (difusión) de cerebro del Caso II que muestra isquemia reciente en territorio de la arteria cerebral media izquierda.
La demostración de alteraciones segmentarias de la motilidad de las paredes del VI contribuye al diagnóstico de esta entidad. Estos trastornos en la motilidad se han reportado en pacientes con lesión neurológica aguda10, 11.
La patogénesis certera del miocardio atontado no se ha establecido, probablemente porque involucra procesos multifactoriales y complejas interacciones intracelulares. Existen tres hipótesis que podrían explicar este fenómeno:
1. Hipótesis del Ca++ y troponina I: La misma implicaría la existencia de una sobrecarga transitoria de Ca++ durante la isquemia y en la fase temprana de la reperfusión; su preciso mecanismo es desconocido. Se postula que durante la isquemia, la célula se ve forzada a realizar metabolismo anaeróbico, con la consiguiente acumulación de protones y disminución del pH intracelular, esto estimula el intercambiador Na+/H+ que expulsa los H+ y aumenta el Na + intracelular, inhibiendo la bomba Na/ K ATPasa. Este aumento del Na+ intracelular modifica la actividad del intercambiador Na+/Ca++ aumentando la concentración de Ca++ intracelular; la sobrecarga de Ca++ activaría las proteasas Ca++ dependientes, que estarían involucradas en la proteolisis de la troponina I. De esta manera se produciría proteolisis parcial de la troponina I, la cual inhibe directamente la unión actinamiosina12, 13. Murphy et al14 describieron por primera vez una disminución de la sensibilidad al Ca++ en las miofibrilllas, manifestada por una disminución en la fuerza máxima y una relativa insensibilidad a la concentración de Ca++ extracelular, modificando el mecanismo fisiológico de la contracción muscular.
El miocardio atontado podría ser visto como una alteración de corto tiempo de la función de los miofilamentos.
2. Especies de Oxígeno reactivo (ROS): A principios de 1980 numerosos investigadores ensayaron esta teoría. La misma sostiene que en la fase temprana de la reperfusión se producirían especies de oxígeno reactivo tales como el anión superoxido, peróxido de hidrógeno, y radicales hidroxilo que alterarían el transporte de calcio a través de la membrana plasmática, lo cual resulta en una sobrecarga de Ca++ intracelular. De esta manera la disfunción contráctil resultaría de la exposición directa a ROS.
3. Genética del atontamiento miocárdico: Algunos autores han encontrado que más del 30% de los genes presentan fenómeno de sobrerregulación en el miocardio atontado, que involucra diferentes mecanismos de sobrevida celular, como: resistencia a la apoptosis, citoprotección y crecimiento celular. Muchos de estos mecanismos han sido implicados previamente en la sobrevida de otros tipos celulares, pero no habían sido descriptos anteriormente en el corazón15. Se ha encontrado la inducción de numerosos genes que no se expresan en el miocardio normal, los cuales participan en el desarrollo y crecimiento de diferentes tipos celulares. Existe un gradiente de respuesta genética relacionado a la reducción de flujo, lo cual muestra que la respuesta nuclear no es un fenómeno de todo o nada sino que es proporcional a la intensidad del estímulo inicial. Los cambios inesperados en los genes y las proteínas podrían ser una nueva aproximación para dilucidar los mecanismos multifactoriales involucrados en el atontamiento y la hibernación miocárdica15.
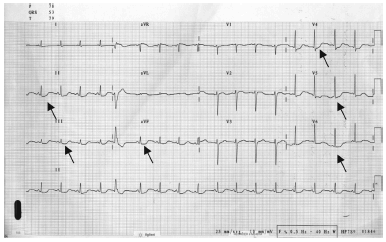
Fig. 4.- Electrocardiograma del Caso II que muestra ondas T negativas e infradesnivel del segmento ST en DII, DIII, aVF1 V4, V5 y V6.
Aproximadamente un tercio de los genes nucleares codificados que fueron regulados en el miocardio isquémico participan en mecanismos de sobrevida celular, como: inhibición de apoptosis, citoprotección, crecimiento celular y estimulación de la translación. Este fenómeno fue más evidente en el subendocardio que en el subepicardio, reflejando que esta respuesta fue proporcional al insulto isquémico15.
Se ha demostrado en modelos experimentales que la administración de adenosina durante la reperfusión protege al miocardio de las alteraciones post-isquémicas sistólicas y del aumento de la rigidez diastólica, sin modificar la relajación isovolumétrica; esta protección estaría directamentemente relacionada con la activación de los receptores A1 purinérgicos.
La actividad simpática aumentada se traduce en dos efectos directos: sobre el pulmón y la fibra miocárdica. A nivel pulmonar la venoconstricción de las venas pulmonares y el aumento de la permeabilidad capilar (mediada por endorfinas, histamina y bradiquininas) genera edema pulmonar por mecanismo mixto (incremento de la presion hidrostática y aumento de la permeabilidad). En la fibra miocárdica la liberación brusca de adrenalina y noradrenalina, por efecto simpático directo o por liberación desde la corteza adrenal, provocará disfunción ventricular izquierda con incremento secundario de la presión capilar pulmonar, contribuyendo con este mecanismo al desarrollo de edema pulmonar.
Los casos clínicos expuestos corresponden a mujeres jóvenes, sin factores de riesgo para enfermedad coronaria, que luego de un infarto cerebral presentaron cambios en el ECG sugestivos de isquemia, elevación de la enzimas cardíacas y alteraciones de la motilidad miocárdica, asociado a edema agudo de pulmón; todos ellos revirtieron al décimo día aproximadamente del insulto inicial.
El interés de esta presentación se basa en la escasa existencia de informes clínicos que mencionen la asociación entre accidente cerebrovascular isquémico, disfunción miocárdica reversible3 y edema de pulmón neurogénico, así como también en la etiología compartida y simultánea de las tres entidades. Nosotros consideramos definir esta entidad como síndrome cerebro-cardiopulmonar.
Bibliografía
1. Heyndrick GR, Millard RW, Mc Ritchie RJ, Maroko PR, Vatner SF. Regional myocardial functional and electrophysiological alterations after brief coronary artery occlusion in conscious dogs. J Clin Invest 1975; 56: 978-85.
2. Mayer SA, Lin J, Homma S, et al. Myocardial injury and left ventricular performance after subarachnoid hemorrhage. Stroke 1999; 30: 780-6.
3. Wang TD, Wu CC, Lee YT. Myocardial stunning after cerebral infarction. Int J Cardiol 1997; 58: 308-11.
4. Davis TP, Alexander J, Lesch M. Electrocardiographic changes associated with acute cerebrovascular disease: a clinical review. Prog Cardiovasc Dis 1993; 36: 245-60.
5. Myers MG, Norris JW, Hachniski VC, Sole MJ. Plasma norepinephrine in stroke. Stroke 1981; 12: 200-4.
6. Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 1999; 79: 609-34.
7. Aschenbrenner R, Bodechtel G. Ueber EKG-Veraenderungen bei Hirntumorkranken. Klin Wochenschr 1938; 17: 298-302.
8. Brouwers PJAM, Wijdicks EFM, Hasan D, et al. Serial electrocardiographic recording in aneurismal subarachnoid hemorrhage. Stroke 1989; 20: 1162-7.
9. Parekh N, Venkatesh B, Cross D, et al. Cardiac troponin I predicts myocardial dysfunction in aneurysmal subarachnoid hemorrhage. J Am Coll Cardiol 2000; 36: 1328-35.
10. Huttemann E, Schelenz C, Chatzinikolaou K, Reinhart K. Left ventricular dysfunction in lethal severe brain injury: impact of transesophageal echocardiography on patient management. Intensive Care Med 2002; 28:1084-8.
11. Dujardin KS, McCully RB, Wijdicks EF, et al. Myocardial dysfunction associated with brain death: clinical, echocardiographic, and pathologic features. J Heart Lung Transplant 2001; 20: 350-7.
12. Mosca SM, Cingolani HE. The Na+/Ca2+ exchanger as responsible for myocardial stunning. Medicina (Buenos Aires). 2001; 61: 167-73.
13. Cingolani HE, Perez NG, Camilion de Hurtado MC. What is Na+/H+ exchanger guilty of in cardiology?Medicina (Buenos Aires). 2000; 60: 709-21.
14. Murphy AM, Kögler H, Georgakopoulos D, et al. Transgenic mouse model of stunned myocardium. Science. 2000; 287: 488-91.
15. Depre C, Tomlinson JE, Kudej RK, et al. Gene program for cardiac cell survival induced by transient ischemia in conscious pigs. Proc Natl Acad Sci U S A. 2001; 98: 9336-41. [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ]
Recibido: 10-05-2005
Aceptado: 3-03-2006

