










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.66 n.5 Buenos Aires sep./oct. 2006
Mutaciones del gen de la Hemocromatosis en donantes de sangre voluntarios y en pacientes con Porfiria cutánea tarda en Chile
Carlos Wolff F1, Rodolfo Armas M1, Jorge Frank2, Pamela Poblete G2
1Departamento de Medicina Occidente, Universidad de Chile, Hospital San Juan de Dios. Santiago, Chile;
2Department of Dermatology, University Hospital Maastricht, The Netherlands
Dirección postal: Dr. Carlos Wolff, Casilla 33052, Santiago, Chile. Fax: (56-2) 681 7414
E-mail: cwfl255@yahoo.es
Resumen
La acumulación de hierro hepático asociada a mutaciones en el gen HFE de la hemocromatosis hereditaria (HH) en los pacientes con porfiria cutánea tarda (PCT) podría tener un papel en la etiología y en la expresión clínica de esta enfermedad. Se estudió la frecuencia de las mutaciones H63D y C282Y en un grupo de pacientes con PCT y se la comparó con la observada en un grupo de donantes voluntarios de sangre. Los pacientes con PCT fueron catalogados como portadores de la forma hereditaria o adquirida de la enfermedad, según presentaran o no mutaciones en el gen uroporfirinógeno decarboxilasa (UROD). El 50% de los pacientes con PCT eran portadores de la forma genética de la enfermedad, porcentaje significativamente mayor que lo informado en otras series. El 23% de los donantes voluntarios de sangre eran portadores de la mutación H63D y 2.4% lo era de la mutación C282Y. Frecuencias similares a lo encontrado por otros autores en población chilena de etnia blanca, en población argentina y española, pero significativamente más alta que lo encontrado en estudios en población aborigen araucana. Esto tiene, probablemente, relación con el predominio de ascendencia española en la población blanca chilena. La frecuencia de mutación en el gen HFE en pacientes con PCT no fue significativamente diferente que la observada en donantes voluntarios de sangre. Tampoco hubo diferencias significativas en la frecuencia de estas mutaciones entre los casos con PCT adquirida respecto de aquellos en que ésta era de origen genético. Los resultados obtenidos no permiten afirmar que exista asociación entre la PCT y la condición de portador de mutaciones del gen HFE de la hemocromatosis hereditaria.
Palabras clave: Hemocromatosis hereditaria; Porfiria cutánea tarda; depósito excesivo de hierro
Abstract
Mutations of hemochromatosis gene in volunteer blood donors and Chilean porphyria cutanea tarda patients. In patients with porphyria cutanea tarda (PCT), hepatic iron accumulation associated to hereditary hemochromatosis (HH) could play a role in the etiology and in the clinical expression of the disease. The H63D and C282Y mutations of the HFE gene frequency were studied in a PCT group of patients and compared with the frequency observed in a group of volunteer blood donors. PCT patients were cataloged as hereditary or acquired PCT carriers, whether or not they presented uroporphyrinogen decarboxilase gene mutations. Fifty percent of PCT patients were carriers of the disease's genetic type. Such percentage is significantly higher than what other authors have previously informed. H63D and C282Y mutations were present in 23% and 2.4% of the volunteer blood donors, respectively. Similar frequencies were informed by others authors in Chilean white ethnic populations, and also in Spaniard and Argentinean populations, but significantly higher than that observed in Chile's Araucanean aboriginal population. Probably the frequency of H63D and C283Y mutations are related to the Spaniard ascendancy dominance of Chile's white ethnic population. The frequency of HFE gene mutations in PCT patients was not different than what was observed in volunteer blood donors. Similarly, there was no statistical difference in the frequency of these mutations among patients with acquired or genetic PCT disease. With the obtained results, it is not possible postulate an association between PCT and the hereditary hemochromatosis of HFE gene mutations carrier conditions.
Key words: Hereditary hemochromatosis; Porphyria cutanea tarda; Iron overload
La porfiria cutánea tarda (PCT) (OMIM 176.100) es la variedad de porfiria más frecuente en todo el mundo, estimándose, por ejemplo, que su prevalencia en España es de alrededor de 1 en 1.000 habitantes1. La PCT se caracteriza por presentar fotosensibilidad y labilidad cutánea debido al aumento en la circulación, acumulación y excreción de uroporfirinas y porfirinas parcialmente decarboxiladas de origen hepático. El aumento de estas porfirinas se debe a la inhibición parcial de la actividad de la uroporfirinógeno decarboxilasa (UROD, EC 4.1.1.37), defecto que puede ser adquirido (PCT esporádica o tipo I, OMIN 176.090) que se expresa exclusivamente en el hígado, o bien de origen genético con transmisión autosómica dominante (PCT familiar o tipo II) en que el defecto de la actividad de la UROD está presente en todos los tejidos, incluida la sangre. La UROD es un polipétido de 42 kDa codificado por un gen único (UROD) de 3kb, constituido por 10 exones, ubicado en el brazo corto del cromosoma 1p34. La identificación de mutaciones en el gen UROD en muestras de sangre permite reconocer el carácter genético de la enfermedad2. La mayoría de los portadores de mutaciones en el gen UROD no expresan el fenotipo clínico debido a la baja penetrancia que tienen estas alteraciones genéticas, expresándose la enfermedad sólo cuando factores adicionales están presentes.
Por su parte, se ha propuesto varios agentes contribuyentes al desarrollo de la PCT esporádica, entre ellos el etanol, los estrógenos, las infecciones virales (hepatitis C y VIH), los hidrocarburos policlorados (hexaclorobenzeno) y la hemodiálisis3.
Los pacientes con PCT frecuentemente tienen acumulación hepática excesiva de hierro, cuya causa es desconocida. Esta siderosis podría contribuir al daño hepático que comúnmente presentan estos pacientes y a que la enfermedad se exprese clínicamente4.
La PCT presenta asociación con otras enfermedades tales como lupus eritematoso, diabetes, alcoholismo, infecciones con los virus B y C de la hepatitis5 y de la inmunodeficiencia adquirida, así como también con la hemocromatosis hereditaria6. Estas asociaciones ocurrirían especialmente en casos de PCT de tipo adquirida o tipo II.
La hemocromatosis hereditaria (HH) es una enfermedad genética debida a la absorción intestinal descontrolada de hierro, el que se deposita anormalmente en diversos tejidos alterándolos anatómica y funcionalmente. La acumulación de Fe en el hígado se produce con mayor intensidad en los hepatocitos de la zona periportal y menor en los del centro del lobulillo. En los casos más graves puede acumularse este mineral en las células reticuloendoteliales, macrófagos y células de conductos biliares. No obstante, el depósito de Fe en células reticuloendoteliales y macrófagos con o sin focos de depósito en hepatocitos, es más propio de sobrecargas de Fe secundarias a otras enfermedades hepáticas7. La HH no es la única condición en que hay acumulación de hierro en el organismo, ésta puede ser ocasionada por una diversidad de condiciones, genéticas o adquiridas, no relacionadas con la absorción intestinal aumentada de Fe y que se conoce como hemosiderosis.
La HH es la enfermedad genética metabólica del adulto más frecuente en la población caucásica, especialmente de origen celta. Uno de cada 20 a 25 individuos caucásicos es portador de una alteración genética asociada a esta enfermedad8. Se expresa sólo en una minoría de éstos, aun siendo homocigotos, dependiendo, entre otros factores, de las pérdidas fisiológicas o patológicas de sangre, aporte de Fe en la dieta, consumo de alcohol, presencia de infecciones crónicas por virus hepatotropos y la presencia de algunas anemias crónicas.
En base a alteraciones clínicas y genéticas, se han identificado 4 tipos de hemocromatosis hereditarias (OMIN data base, Online Mendelian Inheritance in Man Database)9.
Tipo 1: Conocida también como hemocromatosis hereditaria (HH), mal nombre, pues los otros tipos también son hereditarios. Es la variedad más frecuente y su transmisión es autosómica recesiva. Es debida a mutación del gen HFE ubicado en el cromosoma 6p21,3110.
Tipo 2 o forma juvenil: Se distinguen dos subtipos: Subtipo A, cuyo defecto genético se localiza en la porción cromosómica 1q21. Es de transmisión autosómica recesiva, se caracteriza por el depósito tisular de Fe antes de los 30 años de edad, afectando por igual a ambos sexos. Induce la aparición de miocardiopatía e hipogonadismo graves. No se ha identificado el gen implicado, pero se sabe que no es en el HFE. Provisoriamente se le ha denominado HjV. Subtipo B, debida a mutación en el gen ubicado en la porción cromosómica 19q13.1 que codifica para la proteína hepcidina.
Tipo 3: Clínicamente indistinguible de la Tipo 1 pero asociada a mutaciones en el gen que codifica para el receptor de transferrina (TfR-2) ubicado en porción cromosómica 7q22.
Tipo 4: Semejante también a la Tipo 1, causada por mutaciones en el gen SLC40A1, ubicado en la fracción génica 2q32, que codifica la proteína transportadora ferroportina.
En la HH, dos mutaciones de tipo missense del gen HFE son responsables de la mayor parte de los casos de HH. Estas son la C282Y o Cys282Tyr (sustitución de cisteína por tirosina en la posición 282) y la H63D o His63Asp (sustitución de histidina por aspartato en la posición 63). La proteína HFE anormal producto del gen alterado, determina un aumento de la absorción intestinal de Fe como también un aumento de la internalización celular del complejo transferrina-Fe, lo que lleva a un aumento del Fe libre intracelular (no unido a hemosiderina) considerado un agente tóxico por su capacidad de favorecer la generación de radicales libres11.
Aunque la condición de homocigoto para esta mutación es un importante factor de riesgo para la enfermedad, ésta se expresa fenotípicamente sólo en algunos de ellos. Por eso debe diferenciarse la condición de portador del defecto genético de la existencia de la enfermedad clínica.
La mutación H63D tiene una amplia distribución en todo el mundo con mayor prevalencia en la población de origen vasco12. Su presencia tendría significado clínico sólo cuando se acompaña de otras alteraciones genéticas u otras condiciones patológicas como porfiria hepática, hepatitis viral, talasemia.
La condición de heterocigoto compuesto (C282Y/H63D) también es un factor de riesgo para esta enfermedad y, de hecho, está presente en el 3-5% de los casos.
Las personas que son heterocigotos para las mutaciones señaladas, habitualmente son asintomáticas. Sin embargo, cabe destacar que aun cuando no haya expresión clínica de la HH, la presencia de las mutaciones H63D y C282Y, sea como homo o heterocigoto, se asocia a sobrecarga de Fe, que puede tener importancia en quienes padecen enfermedades como cardiopatías, hepatopatías crónicas, artropatías, porfiria cutánea tarda, impotencia e infertilidad, haciendo que éstas sean más graves y de expresión más temprana13. En pacientes con infección por hepatitis por virus C, la proteína HFE mutada puede influir en la progresión de la enfermedad y en la mala respuesta al tratamiento con interferón; por ello se ha recomendado la remoción de Fe en ellos.
La HH es 5 veces más frecuente en los hombres que en las mujeres. También es de aparición más temprana en ellos produciendo las primeras manifestaciones clínicas entre los 30 y los 50 años, lo que ocurre, en cambio, después de los 50 años en las mujeres. Algunas personas pueden presentar síntomas a partir de los 20 años de edad correspondiendo, probablemente, a una variedad juvenil.
El consumo exagerado de alcohol y el antecedente familiar de HH son factores de riesgo para esta enfermedad.
La prevalencia de mutaciones del gen HFE varía según la composición étnica de la población estudiada. Así por ejemplo, la prevalencia de homocigotos (C282Y/C282Y) en población del estado de Utah, EE.UU., es de 1/300, en originarios de la Bretaña de 1/400, y en escoceses de 1/500. En población sueca la prevalencia de sobrecarga de Fe (ferritina y % saturación de la trans-ferrina) sería de 0.5%, lo cual significaría que el 12.8% de la población es portadora de mutaciones del gen HFE.
Diversos autores han planteado que la acumulación de hierro relacionada a las mutaciones C282Y y H63D del gen HFE podría jugar un papel en la etiología o en la expresión clínica de la PCT6, pero las conclusiones han sido contrapuestas. Con el fin de contribuir a comprobar una posible asociación entre PCT y presencia de las mutaciones H63D y C282Y en el gen HFE, estudiamos la frecuencia de estas mutaciones en pacientes portadores de PCT y en un grupo de donantes voluntarios de sangre de un hospital público del área occidente de salud de la ciudad de Santiago.
Materiales y métodos
Se estudió la presencia de las mutaciones H63D y C282Y en el gen HFE en 20 pacientes con PCT -pertenecientes a 16 familias chilenas- 11 en que la enfermedad se consideró de origen genético por presentar mutaciones en el gen UROD14en muestras de sangre y 9 en quienes la PCT se consideró esporádica por no haber identificado mutaciones en el gen UROD. Ninguno de los pacientes estudiados señaló tener un antepasado de origen extranjero en a lo menos 3 generaciones anteriores. El intervalo de edad de los pacientes con PCT al momento del estudio fue 45-83 años, sin diferencia significativa entre aquellos con PCT de origen genético de los que eran portadores de PCT adquirida.
El diagnóstico de PCT se sospechó en base a la presencia de fotosensibilidad cutánea y síntomas cutáneos característicos como vesículas, labilidad, hiperpigmentación, hipertricosis, confirmándose por la excreción urinaria aumentada de uroporfirinas y coproporfirinas y la presencia en heces de isoco-proporfirinas identificadas mediante cromatografía de capa fina4.
El grupo de comparación estaba constituido por donantes voluntarios de sangre del Hospital San Juan de Dios de Santiago de Chile, el que se espera debiera tener una composición étnica semejante a la de los pacientes con PCT incorporados a este estudio. La mutación H63D se buscó en 178 y la C282Y en 82 donantes voluntarios de sangre. Tanto los pacientes con PCT como los donantes voluntarios de sangre, dieron su consentimiento informado para participar en este estudio. El 70.3% del grupo en que estudió la presencia de la mutación C282Y eran hombres y la edad promedio al momento de tomar la muestra de sangre fue de 35.3 ± 9.8 años. Por su parte, el grupo de donantes en que se estudió la mutación H63D tenía una edad promedio de 30.2 ± 7.4 años y el 65.8% eran hombres.
El ADN genómico obtenido de leucocitos de muestras de sangre periférica tomadas en tubos con EDTA, fue aislado mediante procedimientos estándar15.
Las mutaciones del gen HFE se estudiaron mediante la técnica de PCR descrita por Merryweather-Clarke et al.12 y las del gen UROD, siguiendo una estrategia que consistió en amplificar mediante PCR los exones codificantes utilizando primers diseñados para este estudio. Los productos de amplificación fueron sometidos a CSGE (conformation sensitive gel electrophoresis) previamente descrito14, 16. Para confirmar las mutaciones identificadas se realizó análisis de restricción enzimática con endonucleasas de restricción o una combinación de análisis CSGE y secuenciación automática.
El análisis estadístico de los resultados se efectuó utilizando la prueba exacta de Fisher. Se consideró como nivel de significación estadística un valor de p < 0.05.
Resultados
En el grupo de donantes voluntarios de sangre se encontró que 21.3% era portador heterocigoto y 1.7% portador homocigota de la mutación H63D del gen HFE. Por su parte la mutación C282Y se encontró en forma heterocigota en el 2.4% de los casos. No hubo casos de heterocigotos compuestos (Tabla 1). En consecuencia, la frecuencia alélica de H63D es de 12.4% y de 1.2% para C282Y.
TABLA 1.- Frecuencia de genotipos del gen HFE en donantes voluntarios de sangre

En 11 pacientes con PCT diagnosticada clínicamente -pertenecientes a 8 familias- se encontró 6 mutaciones diferentes en el gen UROD, confirmando en ellos el carácter genético de la enfermedad. En los otros 9 pacientes con PCT no se encontró mutaciones en el gen de la URO-D atribuyéndose carácter adquirido a la enfermedad (Tabla 2).
TABLA 2.- Mutaciones en el gen URO-D y genotipos HFE en pacientes con porfiria cutánea tarda
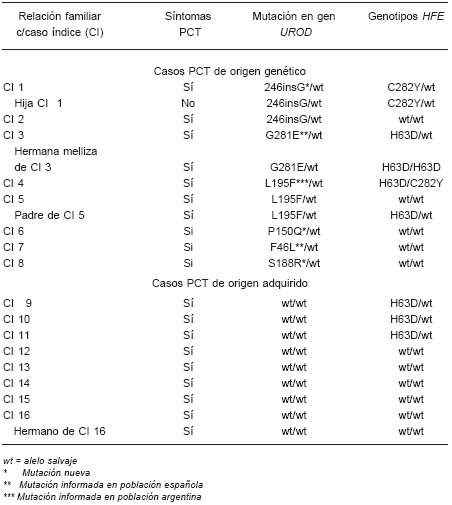
Cinco mutaciones son tipo missense y una de tipo frameshift. Tres mutaciones no han sido informadas previamente en otras poblaciones. Las mutaciones de tipo missense nuevas producen una reducción importante de la actividad UROD evaluada mediante estudios de expresión in vitro14.
Considerando sólo los casos índices, esto es, excluyendo los familiares de los casos índices con PCT, el 50% de los pacientes con PCT de este estudio presenta un origen genético de la enfermedad.
En 9 de los 20 (45%) pacientes con PCT (54% de aquellos en que la PCT era genéticamente dependiente y 33% de los con PCT esporádica) se encontró mutaciones en gen HFE, cuyos genotipos fueron: un homocigoto H63D/H63D, un heterocigoto compuesto C282Y/H63D, 5 heterocigotos H63D/wt y 2 heterocigotos C282Y/wt (Tabla 2).
La frecuencia tanto de casos portadores de la mutación H63D como de la mutación C282Y en pacientes con PCT no fue significativamente diferente (p > 0.05) a la encontrada en el grupo de dadores de sangre. Tampoco hubo diferencia estadísticamente significativa entre la frecuencia de pacientes portadores de mutaciones de HFE al comparar el grupo de pacientes con PCT de origen genético con el grupo en que la PCT era adquirida. Si el análisis se efectúa considerando sólo los casos índice, esto es, excluyendo los familiares de los casos índice, tampoco hubo diferencias estadísticamente significativas en la frecuencia entre casos índice con PCT de origen genético y adquiridos portadores de mutaciones del gen HFE.
La frecuencia alélica, tanto de la mutación H63D como de la mutación C282Y en dadores de sangre (12.4% y 1.2% respectivamente) es similar a las encontradas en el grupo de pacientes con PCT (20.0% y 7.5% respectivamente).
Por su parte, la frecuencia alélica de la mutación H63D en el grupo de pacientes con PCT genética (22.7%) es semejante a la encontrada en el grupo de pacientes con PCT adquirida (16.7%).
Discusión
La alta frecuencia de donantes voluntarios de sangre con mutación H63D (23.0%) con seguridad tiene relación con el gran predominio de ascendencia española de nuestra población6.
La frecuencia alélica tanto de la mutación H63D como de la C282Y encontrada en donantes voluntarios de sangre chilenos, es muy similar a lo informado previamente por Wohllk N. et al.17 en donantes voluntarios de sangre en otro centro hospitalario de Santiago.
La mayor frecuencia del alelo H63D (12.4%) que la del C282Y (1.2%) es semejante a lo informado en población argentina18, brasileña19 y española20, difiriendo a lo encontrado en otros grupos étnicos caucásicos. La frecuencia del genotipo H63D/wt en dadores de sangre chilenos que representan al grupo étnico blanco es significativamente mayor que lo encontrado por Wohllk et al.17 en población araucana, la etnia aborigen mayoritaria de Chile.
La asociación de las mutaciones genéticas propias de la hemocromatosis con la PCT ha sido descrita por diferentes autores, pero con prevalencia muy variable según la composición étnica de la población. En efecto, en pacientes franceses con PCT se encontró que alrededor de 15% eran homocigotas y 15% heterocigotas para la mutación C282Y21. Por su parte, en población sudafricana, 46% de los casos con PCT tenían a lo menos un alelo de la mutación C282Y, 15% era homocigota y 15% heterocigota compuesto con la mutación H63D22. En otro estudio efectuado en sudafricanos con PCT se encontró alta prevalencia de mutaciones C282Y y H63D cuando los pacientes eran de origen europeo, alta prevalencia sólo de la H63D si eran de origen asiático y no se encontró estas mutaciones en los de origen africano6. En población americana, el 65% de los pacientes con PCT resultaron tener también, al menos una mutación para el gen HFE, 29% la C282Y y 47% la H63D.
En este estudio, la frecuencia de mutaciones del gen HFE en pacientes con PCT fue semejante a la observada en el grupo de comparación. Por su parte, la frecuencia de mutaciones del gen HFE tampoco fue estadísticamente diferente entre pacientes PCT tipo I (adquirida) que en tipo II (familiar), señalando con ello que no es posible sostener la existencia de una real asociación entre PCT y HH.
Se sabe que la mutación H63D es de poca expresión fenotípica, lo que explicaría la observación infrecuente de casos de HH en Chile, no obstante la alta frecuencia de ella en el grupo de dadores de sangre. Sin embargo, es posible que los portadores de esta mutación, puedan acumular hierro, aunque sin llegar a presentar HH. Tales pacientes, si además son alcohólicos o infectados por virus de hepatitis C, pueden tener una enfermedad hepática que se exprese con mayor intensidad23. Lo mismo es válido para una mayor expresión de la diabetes por acumulación de hierro en el páncreas.
Llama la atención que en esta serie 50% de los pacientes con PCT presente una alteración genética del gen UROD, ya que en la mayoría de las series estudiadas no sobrepasa el 20%. Autores españoles señalan como muy alta la ocurrencia de este tipo de porfiria, siendo ésta el 37% de su casuística24.
Los resultados obtenidos en este estudio, realizado en pacientes chilenos con PCT, no permiten afirmar la existencia de una asociación entre esta enfermedad, sea de origen genético o adquirido, con la condición de portador de mutaciones en el gen de la hemocromatosis (HFE).
Agradecimientos: La realización de este trabajo se efectuó parcialmente con fondos provenientes de la Fundación de Estudios Biomédicos Avanzados (FEBA) de la Facultad de Medicina de la Universidad de Chile, proyecto 503.
Bibliografía
1. Santos JL, Grandal M, Fontanellas A, Moran MJ, Enríquez de Salamanca R. Prevalence of porphyria cutanea tarda in Madrid and relationship between urine porphyrin and ethanol intake in a multiple linear regression model. Med Clin (Barc) 1996; 107: 614-6.
2. Wolff C, Stella AM, Armas R, Parraguez A, Silva H, Batlle AM. Carácter adquirido de la porfiria cutánea tarda en pacientes infectados con virus C de la hepatitis. Rev Med Chil 1998; 126: 245-50.
3. Bickers Dr, Frank J: The Porphyrias en: Dermatology in General Medicine, Fritzpatrick TB, Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz Si. Editores. 6ª Edición. MacGraw Hill, 2003.
4. Armas R, Wolff C, Krause P, Chana P, Parraguez A, Soto J. Las porfirias hepáticas: Experiencia con 105 casos. Rev Med Chil 1992; 120: 259-66.
5. Wolff C, Armas R, Puig A, Krause P, Náquira N, Parraguez A. Infección por virus C de la hepatitis en pacientes portadores de porfiria. Rev Med Chil 1994; 122: 294-8.
6. Hift RJ, Corrigall AV, Hancock V, Kannemeyer J, Kirsch RE, Meissner PN. Porphyria cutanea tarda: the etiological importance of mutations in the HFE gene and viral infection is population-dependent. Cell Mol Biol (Noisyle-Grand) 2002; 48: 853-9.
7. Nash S, Marconi S, Sikorska K, Naeem R, Nash G. Role of liver biopsy in the diagnosis of hepatic iron overload in the era of genetic testing. Am J Clin Pathol 2002; 118: 73-81.
8. Harrison SA, Bacon BR Hereditary hemochromatosis: update 2003. J Hepatol 2003; 38: 14-23.
9. Pietrangelo A. Hereditary Hemochromatosis - A new look at an old disease. N Engl J Med 2004; 350: 2383-97.
10. Feder JN, Gnirke A, Thomas W. et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399-408.
11. Andrews NC, Levy JE: Iron is hot: an update on the pathophysiology of hemochromatosis. Blood 1998; 92: 1845.
12. Merryweatherclarke AT, Pointon JJ, Shearman JD, et al. Global prevalence of putative haemochromatosis muta-tions. J Med Genet 1997; 34: 275-8.
13. Pietrangelo A. Hemochromatosis. Gut 2003; 52 (suppl II): ii23-ii30.
14. Poblete-Gutiérrez P, Mendez M, Wiederholt T, et al. The molecular basis of porphyria cutanea tarda in Chile: Identification and func-tional characterization of mutations in the uroporphyrino-gen decarboxylase gene. Exp Dermatol 2004; 13: 372-9.
15. Sambrook J, Fritsch EF, Maniatis T. (eds.): Molecular cloning: A laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, 1989.
16. Ganguly A, Rock MJ, Prockop DJ. Conformation-sensitive gel electrophoresis for rapid detection of single-base differences in double-stranded PCR products and DNA fragments: Evidence for solvent-induced bends in DNA heteroduplexes. Proc Natl Acad Sci USA 1993; 90: 10325-9.
17. Wohllk N, Zapata R, Acuña M, Reyes H. HFE gene mutations in Chile. Ann Intern Med 2003; 139: 708-9.
18. Méndez M, Sorkin L, Rossetti MV, et al. Familial porphyria cutanea tarda: characterization of seven novel uroporphyrinogen decarboxylase mutations and frequency of common hemochromatosis alleles. Am J Hum Genet 1998; 63: 1363-75.
19. Agostinho MF, Arruda VR, Basseres DS, et al. Mutation analysis of the HFE gene in Brazilian populations. Blood Cells Mol Dis 1999; 25: 324-7.
20. Sánchez M, Bruguera M, Bosch J, Rodes J, Ballesta F, Oliva R. Prevalence of the Cys282Tyr and His63Asp HFE gene mutations in Spanish patients with hereditary hemochromatosis and in controls. J Hepatol 2000; 32: 362-3.
21. Chiaverini C, Halimi G, Ouzan D, Halfon P, Ortonne JP, Lacour JP. Porphyria cutanea tarda, C282Y, H63D and S65C HFE gene mutations and hepatitis C infection: A study from Southern France. Dermatology 2003; 206: 212-6.
22. McCrossin I. Porphyria cutanea tarda in south-east New South Wales. Australas J Dermatol 2002; 43: 285-8.
23. Bulaj ZJ, Phillips JD, Ajioka RS, et al. Hemochromatosis genes and other factors contributing to the patogenesis of porphyria cutanea tarda. Blood 2000; 95: 1565-71.
24. Cruz-Rojo J, Fontanellas A, Moran-Jiménez MJ, et al. Precipitating/aggravating factors of porphyria cutanea tarda in Spanish patients. Cell Mol Biol 2002; 48: 845-52. [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ]
Recibido: 3-01-2006
Aceptado: 3-05-2006

