










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.66 n.5 Buenos Aires sep./oct. 2006
Accidente cerebro-vascular en la enfermedad de Fabry. Algo más que una simple estenosis
Juan Manuel Politei
Servicio de Neurología, Hospital Juan A. Fernández, Buenos Aires
Dirección postal: Dr. Juan Manuel Politei: Servicio de neurología, Hospital General de Agudos Juan A. Fernández, Cerviño 3356, 1425 Buenos Aires, Argentina. Fax: (54-11) 4801-9349
E-mail: jpolitei@hotmail.com
Resumen
Se analiza la evidencia existente a la fecha sobre los mecanismos fisiopatológicos que pueden generar accidentes cerebrovasculares en la enfermedad de Fabry. Esta entidad es el resultado de la deficiencia de a-galactosidasa A, lo que resulta en depósito patológico de glicoesfingilípidos en distintas poblaciones celulares. Asociados a la insuficiencia renal y cardíaca, los accidentes cerebrovasculares pueden derivar en la muerte de los pacientes. Durante mucho tiempo el único mecanismo generador de daño vascular informado fue la oclusión vascular por depósito lipídico a nivel endotelial. En la actualidad se describen otros mecanismos. El advenimiento de la terapia de reemplazo enzimático ha generado gran expectativa en cuanto la posibilidad de reversión de estos mecanismos. Si bien la evidencia es escasa y son necesarios más estudios a largo plazo, algunos informes demuestran que luego de meses, el tratamiento ha logrado revertir algunos de los mecanismos implicados.
Palabras clave: accidente cerebrovascular, enfermedad de Fabry, terapia de reemplazo enzimático
Abstract
Stroke in Fabry disease. More than a simple stenosis. The objective is to analyze the updated evidence on the physiopathological mechanisms that can generate cerebrovascular damage in Fabry disease. Fabry disease is the result of the deficiency of a-galactosidasa A, which causes pathological storage of glycosphingolipids, in different cells. Associated to renal and cardiac insufficiency, cerebrovascular complications can derive in the death of the patients. During a long time the only reported mechanism was the vascular occlusion by deposit of lipids at endothelial level. At the present time, other mechanisms are postulated. The arrival of enzyme replacement therapy has generated great expectation on the possibility of reversion of these alterations. Although the evidence is scarce and more long term studies are necessary, some reports demonstrate that after months, the treatment has managed to revert some of the mechanisms involved.
Key words: stroke, Fabry disease, enzyme replacement therapy
La enfermedad de Fabry (EF) es el resultado de la deficiencia de a-galactosidasa A, lo que resulta en depósito patológico de glicoesfingolípidos, predominantemente globotriaosilceramida (Gl3). Esta entidad, de herencia ligada al cromosoma X, tiene una incidencia de 1/40.000 nacidos vivos1. El depósito de Gl3 se puede observar en células endoteliales, periteliales, neuronas, podocitos, cardiomiocitos, etc2. Los primeros síntomas se expresan durante la niñez con dolor distal de tipo neuropático en los cuatro miembros e hipohidrosis, asociado a lesiones cutáneas conocidas como angioqueratomas. Durante la adolescencia se agrega depósito de Gl3 en córnea (córnea verticilada), nuevas manifestaciones disautonómicas, fatiga y disminución de la capacidad auditiva para, llegada la adultez, desarrollar insuficiencia renal y cardíaca, como también accidentes cerebrovasculares3. Estas últimas complicaciones llevan a la muerte del 50% de los pacientes a los 41 años de vida. Desde hace aproximadamente 4 años la EF ha dado un giro significativo debido a la posibilidad de terapia de reemplazo enzimático4, 5.
El objetivo de este artículo es analizar la evidencia existente a la fecha sobre los nuevos mecanismos fisiopa-tológicos que pueden generar accidentes cerebrovasculares en la EF.
Mecanismos generadores de accidentes cerebrovasculares
Desde las primeras descripciones en 1898 por Anderson y Fabry6, 7, hasta pocos años atrás, el compromiso cerebrovascular en la EF era explicado por la isquemia generada por la oclusión de la luz vascular secundaria al depósito endotelial de glicoesfingolípidos. Esta hipótesis era sostenida por los hallazgos de autopsia y neuro-imágenes8. En la actualidad, las descripciones realizadas por medio de angiografía digital, ecografía doppler, tomografía por emisión de positrones y pletismografía entre otras técnicas, han podido establecer nuevos mecanismos implicados en la generación de accidentes cerebrovasculares (Tabla 1).
TABLA 1.- Mecanismos generadores de accidente cerebro vascular

Compromiso de la pared vascular
Las descripciones angiográficas y de tomografía demostraron la presencia de engrosamiento y dolicoectasia vascular, asociadas a estenosis luminar. Si bien estos hallazgos se podían ver en ambos territorios cerebrovasculares, eran predominantes en el territorio posterior8. Posiblemente una de las explicaciones para esto sea que los sectores distales al nacimiento de las arterias cerebrales posteriores carecen de receptores catecolaminérgicos con la consecuente disminución del tono simpático.
La presencia de dolicoectasia intracraneana ha llevado a la descripción de complicaciones no isquémicas, como hidrocefalia por impacto vascular sobre el tercer ventrículo9, oculoparesias por compresión de nervios craneanos10, así como también neuralgia trigeminal11, paresia del hipogloso9, etc.
El depósito de Gl3 no sólo compromete al endotelio, sino también a la capa de músculo liso de la pared arterial, como se demuestra por medio de anatomía patológica1.
La elasticidad y el tono de la pared vascular son los resultados de una matriz extracelular compuesta por colágeno, elastina, glicosaminoglicanos y proteoglicanos12. Las células musculares incrementan la síntesis de éstas moléculas en respuesta al aumento o redistribución del tono en la pared arterial13, 14. El depósito de Gl3 interfiere en la producción de la matriz extracelular, con pérdida de la integridad estructural de la pared arterial, llevando a una gradual dilatación y formación de aneurismas. Por otro lado, se conoce que las proporciones de los componentes de la matriz extracelular no son similares en las diferentes arterias. Las arterias intracraneanas presentan menor cantidad de elastina y son proporcionalmente más delgadas que las arterias extracraneanas15, 16. Esta diferencia es importante para explicar la presencia de dolicoectasia sólo en arterias intracraneanas.
Compromiso endotelial
Evaluación del sistema vascular periférico
Las células endoteliales intervienen en la regulación del tono arterial por medio de la síntesis de sustancias constrictoras y dilatadoras17. Uno de los factores mejor estudiados es el óxido nítrico (ON)18; estando otros (como el factor hiperpolarizante derivado del endotelio) también implicados. Altarescu et al, informan una exagerada vasodilatación dependiente del endotelio, por vías no relacionadas al ON, al evaluar el flujo sanguíneo en venas del antebrazo con pletismografía19. Para demostrarlo comparó la respuesta del flujo sanguíneo luego de la infusión de acetilcolina (generador de dilatación dependiente del endotelio) y de nitroprusiato de sodio (generador de dilatación por reacción directa con el músculo liso vascular); ambas drogas antes y después de un inhibidor de la síntesis de ON como la N-G-monometil/arginina (L-NMMA); en un grupo de pacientes con EF y en un grupo control. En los pacientes se generó un aumento del flujo significativamente superior en comparación con el grupo control, con la infusión de acetilcolina, aun luego de la L-NMMA. Los resultados no fueron significativos luego de la infusión de nitroprusiato de sodio. Debido a que el compromiso de las fibras simpáticas podría interferir con la respuesta vascular, se midieron los niveles de adrenalina y noradrenalina sérica. Los valores fueron similares en los dos grupos, por lo que se consideró que la inervación simpática vascular estaba intacta.
En relación a este último punto, Stemper et al., al demostrar hipoperfusión en el antebrazo en reposo y luego de la isquemia (por medio de pletismografía), asociado a hiperperfusión cutánea post-isquemia20, concluye que la hipoperfusión inicial es el resultado del depósito lipídico endotelial, pericítico y muscular liso y que la ausencia de vasocontricción post-isquemia sería por compromiso de fibras simpáticas intracutáneas.
Evaluación del sistema vascular central
Para la evaluación del flujo sanguíneo cerebral (FSC), Moore et al utilizaron tomografía por emisión de posi-trones, mediciones plasmáticas de nitrato y nitritos y tinción de nitrotirosina en piel y tejido cerebral21. Los hallazgos fueron compatibles con un aumento del FSC regional en los pacientes en comparación con los controles, a predominio de áreas posteriores22. La inmunohistoquímica para nitrotirosina mostró una extensa positividad en vasos dérmicos y cerebrales en los pacientes. Los niveles de nitrato plasmático (aunque no en forma significativa) fueron inferiores en los pacientes; esto indicaría una alteración de la síntesis de ON con aumento de su formación, lo que lleva a mayor generación de radicales libres. Los peroxinitritos resultantes generan vasodilatación y resistencia a la vasoconstricción por mediadores humorales. Se ha notificado mejoría del FSC luego de la infusión intravenosa de vitamina C, lo que confirma la presencia de procesos oxidativos23.
Compromiso del metabolismo central
Debido a que los resultados de autopsias muestran compromiso vascular asociado a depósito lipídico en gran cantidad de neuronas, se ha propuesto que los cambios en el metabolismo celular podrían traer aparejado cambios en el FSC. Para evaluar si el aumento del FSC regional era secundario a un aumento en el metabolismo celular, se realizó un estudio con tomografía por emisión de positrones que midió el consumo de glucosa y el FSC en forma global y regional24. Los resultados demostraron que la utilización de glucosa cerebral (UGC) global no era diferente entre los controles y los pacientes. Sin embargo, el UGC regional era menor en las áreas con mayor riesgo de isquemia y aumento del FSC. Esto demostraría que el aumento del FSC no es por una mayor demanda (aumento del metabolismo celular) sino, en parte, por la falta de control autonómico del sistema cerebro-vascular. Otra hipótesis que explica esta disociación, es que ante la insuficiencia metabólica neuronal, se liberarían distintos mediadores (como la adenosina) que al aumentar el FSC actuarían como protectores de la isquemia resultante25. La insuficiencia metabólica sería secundaria a microangiopatía oclusiva por depósito lipídico, aunque en contraposición, algunos resultados de autopsia no han encontrado esta eventualidad26.
De lo antedicho se puede concluir que el desarrollo de lesiones de la sustancia blanca es consecuencia de una cadena de eventos que se pueden resumir en la Fig. 1.
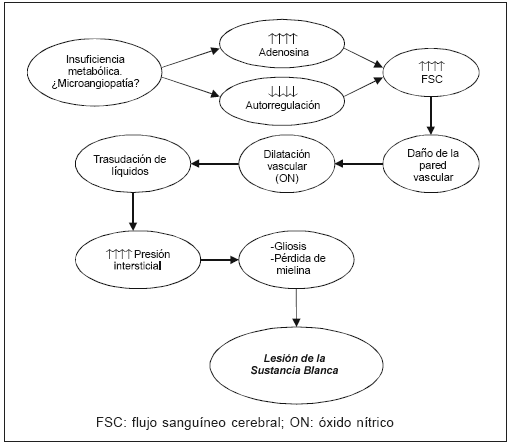
Fig. 1.- Cascada de mecanismos generadores de accidentes cerebrovasculares
Compromiso del control autonómico de la regulación cerebrovascular
El eco-Doppler transcraneano es una forma no invasiva indirecta de evaluar el flujo cerebrovascular en reposo y la capacidad de regulación por medio de distintas técnicas. Los resultados reportados a la fecha son dispares. Por un lado, Moore et al, informan velocidades de flujo incrementadas en los pacientes al comparar con grupos controles, así como también los índices de pulsatilidad y de resistencia27. Por otro, Hilz et al; encuentran reducción de las velocidades de flujo, asociado a falta de autorregulación vascular cerebral28. En nuestra experiencia los resultados obtenidos son similares a los de Hilz (artículo en revisión). Los mecanismos que regulan el tono vascular dependen de factores miogénicos, endoteliales y neurogénicos29. La reducción del control vasomotor simpático puede ser atribuida al depósito de Gl3 en neuronas autonómicas centrales y periféricas1, lo que genera otro mecanismo de hipoflujo cerebral en un territorio predispuesto por la estenosis vascular.
Activación endotelial e hipercoagulabilidad
El aumento en la incidencia de trombosis arteriales y venosas en los pacientes con EF fue descripto por Utsumi et al30. Se ha demostrado que la exposición de los leucocitos a los glicoesfingolípidos de la pared vascular resulta en un aumento en la expresión de moléculas de adhesión en el endotelio31. De Graba et al, informan la medición de la concentración plasmática de diferentes marcadores de activación endotelial, encontrando un aumento significativo en los niveles de moléculas de adhesión intercelular 1 soluble, molécula de adhesión celular vascular 1 soluble, P selectina e inhibidor del activador del plasminógeno (PAI) y una disminución en la concentración de trombomodulina (TM)32. La disminución de la TM no permite la activación de la proteína C trombinamediada; y el aumento del PAI disminuye las propiedades trombolíticas del endotelio33. Por otro lado, los glicoesfingolípidos estimulan la expresión de CD11/CD18 en leucocitos34, lo que lleva a un aumento de la adhesión a la pared vascular y a la generación de un estado procoagulante35.
Demuth et al, informan aumento en los niveles de homocisteína plasmática36. La homocisteína tiene efectos neurotóxicos como resultado de la inhibición de reacciones de metilación y al exceso de producción de aminoácidos que contienen sulfuros37. Otros autores no pudieron reproducir estos hallazgos19.
Por lo expuesto anteriormente, se ha demostrado que la estenosis vascular por depósito de glicoesfingolípidos a nivel endotelial es solo uno de los mecanismos que pueden resultar en un accidente cerebrovascular en los pacientes con EF.
A la fecha existen pocos reportes que demuestren la reversibilidad de estos mecanismos con la terapia de reemplazo enzimático; entre ellos se destaca el trabajo de Chenevard et al, que luego del tratamiento evidencia cambios en la pared vascular38. Otros autores han mostrado mejoría del FSC y reversión de la hiperperfusión cerebral mediante tomografia por emisión de positrones y eco-Doppler transcraneano21, 27.
Serán necesarios estudios de seguimiento a largo plazo para confirmar la reversión completa de estos mecanismos.
Agradecimientos: A Genzyme Corp. Argentina, por el aporte técnico brindado al centro de diagnóstico y tratamiento para la enfermedad de Fabry del Hospital Juan A. Fernández.
Bibliografía
1. Desnick RJ, Ioannou YA, Eng CM. a-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic bases of Inherited Disease. 8th ed. New York: MacGraw-Hill; 2001: 3733-74.
2. Kint JA. Fabry's disease: alpha-galactosidase deficiency. Science 1970; 167: 1268-9.
3. MacDermot Kd, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001; 38: 750-60.
4. Schiffmann R, Kopp JB, Austin HA 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001; 285: 2743-9.
5. Eng CM, Gurron N, Wilcox WR, et al. Safety and efficacy of recombinant human a-galactosidase A-replacement therapy in Fabry's disease. N Engl J Med 2001; 345: 9-16.
6. Anderson W. A case of angiokeratoma. Br J Dermat 1898; 1: 113-7.
7. Fabry J. Ein Beitrag zur Kenntnis der Purpura haemorrhagic a nodularis (Purpura papulosa haemorrhagica Hebrae). Arch Dermatol Syph 1898; 43: 187-200.
8. Mitsias P, Levine SR. Cerebrovascular complications of Fabry's disease. Ann Neurol 1996; 40: 8-17.
9. Maisey DN, Cosh JA. Basilar artery aneurysm and Anderson-Fabry disease. J Neurol Neurosurg Psychiatry 1980; 43: 85-7.
10. De Groot WP. Angiokeratoma corporis diffusum Fabry. Dermatológica 1964; 128: 321-49.
11. Morgan SH, Rudge P, Smith SJM, et al. The neurological complications of Anderson-Fabry disease (a-galactosidase A deficiency)- investigation of symptomatic and presymptomatic patients. Q J Med 1990; 75: 491-504.
12. Dobrin PB. Mechanical propierties of arteries. Physiol Rev 1978; 58: 397-460.
13. Clark JM, Glaskov S. Transmural organization of the arterial media. The lamellar unit revisited. Arteriosclerosis 1985; 5: 19-34.
14. Leung DY, Glaskov S, Mathews MB. Cyclic stretching stimulates synthesis of matrix components by arterial smooth muscle cells in vitro. Science 1976; 191: 475-7.
15. Rodbard S. Negative feedback mechanisms in the architecture and function of the connective and cardiovascular tissues. Perspect Biol Med 1970; 13: 507-27.
16. Stehbens WE. Pathology of cerebral blood vessels. St Louis: Mosby, 1972: 60-2.
17. Vane JR, Anggard EE, Botting RM. Regulatory functions of the vascular endothelium. N Engl J Med. 1990; 323: 27-36.
18. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980; 288: 373-6.
19. Altarescu G, Moore DF, Pursley R, et al. Enhanced endothelium-dependent vasodilation in Fabry disease. Stroke 2001; 32: 1559-62.
20. Stemper B, Hilz MJ. Postischemic cutaneous hyperperfusion in the presence of forearm hypoperfusion suggests sympathetic vasomotor dysfunction in Fabry disease. J Neurol 2003; 250: 970-6.
21. Moore DF, Scott LT, Galdwin MT, et al. Regional cerebral hiperperfusion and nitric oxide pathway dysregulation in Fabry disease: reversal by enzyme replacement therapy. Circulation 2001: 104; 1506-12.
22. Moore DF, Herscovitch P, Schiffmann R. Selective arterial distribution of cerebral hyperperfusion in Fabry disease. J Neuroimaging 2001; 1: 303-7.
23. Moore DF, Ye F, Brennan ML, et al. Ascorbate decreases Fabry cerebral hyperperfusion suggesting a reactive oxygen species abnormality: an arterial spin tagging study. J Magn Reson Imaging. 2004; 20: 674-83.
24. Moore DF, Altarescu G, Barker WC, Patronas NJ, Hers-covitch P, Schiffmann R. White matter lesions in Fabry disease occur in 'prior' selectively hypometabolic and hyper-perfused brain regions. Brain Res Bull 2003; 62: 231-40.
25. Ransom BR, Waxman SG, Fern R. Molecular pathophy-siology of white matter anoxiv/ischemic injury, in: H. Barnett, JP Mohr, BM Stein, FM Yatsu (Eds.), Stroke, Churchill-Livingstone, Philadelphia,1998, pp. 101-9.
26. Kahn P. Anderson-Fabry disease: a histopathological study of three cases with observations on the mechanism of production of pain. J Neurol Neurosurg Psychiatry 1973; 36: 1053-62.
27. Moore DF, Altarescu G, Ling GSF, et al. Elevated cerebral blood flow velocities in Fabry disease with reversal after enzyme replacement. Stroke 2002: 33; 525-31.
28. Hilz MJ, Kolodny EH, Brys M, Stemper B, Haendl T, Marthol H. Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. J Neurol 2004: 251; 564-70.
29. Ursino M. Mechanisms of cerebral blood flow regulation. Crit Rev Biomed Eng 1998; 18: 255-88.
30. Utsumi K, Yamamoto N, Kase R, et al. High incidence of thrombosis in Fabry's disease. Intern Med 1997; 36: 327-9.
31. Scalia R, Appel JZ 3rd, Lefer AM. Leukocyte-endothelium interaction during the early stages of hypercholesterolemia in the rabbit: role of P-selectin, ICAM-1, and VCAM-1. Arterioscler Thromb Vasc Biol 1998; 18: 1093-100.
32. DeGraba T, Azhar S, Dignat-George F, et al. Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol 2000; 47: 229-33.
33. Stern DM, Kaiser E, Nawroth PP. Regulation of the coagulation system by vascular endothelial cells. Hae-mostasis. 1988; 18: 202-14.
34. Arai T, Bhunia AK, Chatterjee S, Bulkley GB. Lactosy-lceramide stimulates human neutrophils to upregulate Mac-1, adhere to endothelium, and generate reactive oxygen metabolites in vitro. Circ Res 1998; 82: 540-7.
35. Marx N, Neumann FJ, Zohlnhofer D, Dickfeld T, Fischer A, Heimerl S. Enhancement of monocyte procoagulant activity by adhesion on vascular smooth muscle cells and intercellular adhesion molecule-1-transfected Chinese hamster ovary cells. Circulation 1998; 98: 906-11.
36. Demuth K, Germain DP. Endothelial markers and homocysteine in patients with classic Fabry disease. Acta Paediatr Suppl 2002; 91: 57-61.
37. Parnetti L, Bottiglieri T, Lowenthal D. Role of homocysteine in age-related vascular and non-vascular diseases. Aging (Milano). 1997; 9: 241-57.
38. Chenevard R, Hurlimann D ,Widmer U, Ruschitzka F, Noll G. Effect of enzyme replacement therapy on intima media thickness and endothelial function in fabry disease. JACC 2004, (suppl 2); 43: 470.
Recibido: 22-11-2005
Aceptado: 24-05-2006

