










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.67 n.1 Buenos Aires ene./feb. 2007
Síndrome hemofagocítico reactivo en pacientes graves. Comunicación de 4 casos
María Mercedes Kleinert1, Gonzalo Garate (H)2, Javier Osatnik1, Juan Cicco2, Bárbara Hunter1, Elisa J. Soria1
1Servicio de Terapia Intensiva,
2Servicio de Hematología, Hospital Alemán, Buenos Aires
Dirección Postal: Dra. María Mercedes Kleinert, Servicio de Terapia Intensiva, Hospital Alemán, Pueyrredón 1640, 1118 Buenos Aires, Argentina. Fax: (54-11) 4827-7014
e-mail: sterapiaintensiva@hospitalaleman.org
Resumen
El síndrome hemofagocítico reactivo, o linfohistiocitosis hemofagocítica secundaria, comprende un grupo numeroso de enfermedades, muchas de ellas de causa infecciosa, caracterizado por hemofagocitosis con citopenia de al menos dos de las tres series sanguíneas, aumento de los niveles de citoquinas y de la ferritina sérica. El cuadro clínico comprende manifestaciones inflamatorias sistémicas semejantes a la sepsis, entidad muy frecuente en las unidades de Terapia Intensiva, y posee elementos fisiopatológicos en común con ella. Proponemos mediante la presentación de cuatro casos clínicos, considerar al síndrome hemofagocítico reactivo como una entidad frecuente en los pacientes graves, con pruebas sencillas para orientar a qué pacientes realizar un procedimiento diagnóstico. Por último actualizamos los tratamientos específicos conocidos.
Palabras clave: Síndrome hemofagocítico; Linfohistiocitosis hemofagocítica; Hemofagocitosis; Ferritina
Abstract
Reactive hemophagocytic syndrome in critical care patients. Report of 4 cases. Reactive hemophagocytic syndrome or hemophagocytic lymphohistiocytosis comprises a variety of disorders, many of them associated with infection. It is characterized by hemophagocytosis, with cytopenia involving at least two cellular lines, increase in cytokines and serum ferritin. The clinical course resembles sepsis, sharing similar physiopathological features. We propose that hemophagocytic syndrome is an underdiagnosed entity in the critical care setting; simple tests aid to identify which patients should undergo diagnostic procedures. We discuss current therapeutic approaches.
Key words: Hemophagocytic syndrome; Hemophagocytic lymphohistiocytosis; Hemophagocitosis
El síndrome hemofagocítico reactivo (SHR) o linfohistiocitosis hemofagocítica (LHH) secundaria es una entidad clínico-patológica caracterizada por la proliferación sistémica de macrófagos benignos con prominente actividad hemofagocitaria. El SHR fue descripto por Chandra y col1 en 1975 en la población adulta, pero fueron Risdall y col2 en 1979 quienes lo caracterizaron adecuadamente distinguiéndolo de la variante histiocitosis maligna infantil. Se informan 4 casos de SHR en pacientes internados en nuestra unidad de Terapia Intensiva de adultos (UTI) desde el año 2001 al 2003 y una reseña de aspectos clínicos, fisiopatológicos y terapéuticos.
Casos clínicos
Caso N° 1: Un varón de 75 años portador de un cáncer de próstata avanzado ingresó por un cuadro de shock séptico que requirió drogas vasoactivas y asistencia respiratoria mecánica (ARM). El score Apache II fue de 22. Los hemocultivos, el urocultivo y el material proveniente de una celulitis de miembro superior izquierdo fueron positivos para Escherichia coli. Recibió tratamiento antibiótico adecuado a su aislamiento. Tuvo alteraciones en las pruebas hepáticas con incremento de la bilirrubina a 1,82 mg/dl, hipoglucemia de 40 mg/dl, plaquetopenia de 39.000/mm3, tiempo de protrombina del 40%, tiempo parcial de tromboplastina de 50 seg y fibrinógeno de 200 mg/dl. Requirió una técnica de diálisis extracorpórea por oligoanuria y cifras de urea y creatinina en aumento. A los 9 días de su ingreso persistió con citopenia en las tres líneas: hemoglobina (Hb):7g/l; leucocitos: 3.700/mm3; plaquetas: 22.000/mm3 y episodios febriles. Se le transfundieron plaquetas los dias 3 y 10. Una punción de médula ósea (PAMO) demostró un aumento de macrófagos de aproximadamente un 5% a 8% con fagocitosis de elementos formes, principalmente eritrocitos. Se le administró gammaglobulina en dosis de 400 mg/kg /día por 5 días. Recuperó el nivel adecuado plaquetario al 6° día. Tuvo una neumonía asociada a respirador en el dia 10 de ARM por Stafilococcus aureus meticilino resistente. La infección del miembro superior requirió limpiezas quirúrgicas y no evolucionó satisfactoriamente. Falleció a los 28 días, luego de haberse acordado con la familia la suspensión de las medidas de sostén.
Caso N° 2: Un varón de 62 años ingresó por insuficiencia respiratoria en el día 20 post transplante autólogo de médula ósea por mieloma múltiple. El score Apache II de ingreso fue 33. Requirió ARM y drogas vasoactivas por shock. El paciente tenía un cociente PaO2/FiO2 de 86, opacidades bilaterales en la radiografía de tórax y el lavado broncoalveolar (BAL) mostró un crecimiento marginal de Pseudomona aeruginosa. Los cultivos y las serologías para virus herpes y citomegalovirus fueron negativos. Recibió tratamiento antibiótico adecuado al aislamiento. A las dos semanas de su ingreso presentó fiebre y empeoramiento de las imágenes radiológicas. Un nuevo BAL fue hemático y no mostró crecimiento bacteriano. El paciente tenía citopenias de las tres series: Hb: 7 g/l; recuento de leucocitos: 600/mm3; plaquetas: 39.000 /mm3, luego de haberse recuperado de la toxicidad del régimen quimioterápico del transplante. Se le realizó una PAMO que mostró un aumento de macrófagos en un 10% con hemofagocitosis (Fig. 1). Se le administró metilprednisona a una dosis de 1g/día. Evolucionó con episodios repetidos de hemorragia alveolar y fiebre. Las citopenias no mejoraron. Se repitieron cultivos que siempre fueron negativos, a excepción de un BAL que evidenció el crecimiento siginificativo de una Pseudomona aeruginosa multirresistente. Falleció a los 64 días del ingreso con falla multiorgánica.
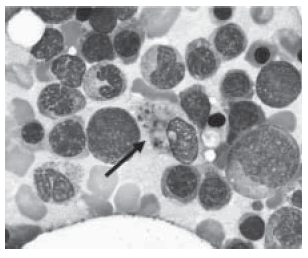
Fig. 1.- Aspirado de médula ósea en May Grunwald Giemsa, vista en 100X. La flecha señala un macrófago con partículas fagocitadas en su citoplasma y restos nucleares. Aparte se observan eritroblastos en distintos estadios de maduración, linfocitos, cayados y monocitos.
Caso N° 3: Un paciente varón de 67 años ingresó por insuficiencia respiratoria secundaria a neumonía. Tenía antecedente de un tumor cerebral operado con posterior tratamiento radiante y quimioterápico. Evolucionó con shock y requerimiento de ARM. Se realizó monitoreo hemodinámico invasivo con valores compatibles con falla cardíaca aguda. Recibió noradrenalina y dobutamina. Por registros febriles se realizó un BAL que desarrolló Acinetobacter baumanii. Se lo medicó con meropenem. Al tercer día de internación necesitó hemodiálisis por fallo renal oligoanúrico. Al 7° día presentó una disminución paulatina de las cifras de plaquetas y anemia, llegando el día 15° a 54.000 plaquetas/mm3 con Hb 7g/l y recuento de leucocitos de 2.000/mm3. Desarrolló fiebre persistente y opacidades bilaterales en la Rx de tórax. Un BAL nuevamente desarrolló Acinetobacter baumanii. La ferritina era 2177 ng/ml (valores normales: hombres 30-400 ng/ml) y los triglicéridos 114 mg/dl. Una PAMO evidenció aumento de un 10% de los macrófagos con hemofagocitosis. Se administró gammaglobulina 400 mg/kg/día por 5 días con recuperación de las tres series y desaparición de la fiebre. El día 26 reapareció el shock que fue refractario al tratamiento vasopresor. Falleció el día 27 de internación.
Caso N° 4: Un paciente de 24 años ingresó por deterioro del sensorio, dolor abdominal, náuseas y vómitos en el 40° día post transplante renal de donante vivo relacionado, por uropatía obstructiva, con reflujo vesicoureteral crónico bilateral. Recibía prednisona 34 mg/día y micofenolato mofetil 500 mg/día como tratamiento inmunosupresor. El score Apache II de ingreso fue 13. El paciente tenía tendencia al sueño, un exantema maculopapuloso descamativo en tronco y el examen abdominal revelaba dolor en hipocondrio y flanco derechos sin reacción peritoneal. Los análisis de laboratorio mostraron Hb:15 g/l; recuento de leucocitos: 6.900 /mm3; plaquetas: 70.000/mm3; creatinina: 1.56 mg/dl; BT: 0.60 mg/dl; GOT: 186 U/l; GPT: 244 U/l; natremia: 121 mEq/l; potasemia: 3.6 mEq/l, LDH: 709 U/l; tiempo de protrombina: 100%; dímero D: negativo. La radiografia de tórax fue normal. A las 48 h la Hb había descendido a 7 g/l, el recuento de leucocitos a 1800 /mm3 y las plaquetas a 49.000/mm3. El paciente presentó fiebre mayor a 38 °C. Los cultivos de sangre y orina fueron negativos. Una antigenemia para CMV fue positiva por lo que se inició tratamiento con ganciclovir. Debido a la persistencia de citopenias en las tres líneas, se efectuó una PAMO que mostró 15 a 20% de histiocitos con hemofagocitosis. Presentaba ferritina: 3517 ng/ml y triglicéridos: 234 mg/dl. Se inició gammaglobulina endovenosa por 5 días. Al 5° dia de tratamiento, continuaba con fiebre y apareció expectoración hemoptoica y opacidades bilaterales en la radiografía de tórax. Requirió ARM por insuficiencia respiratoria. Un BAL fue negativo para gérmenes. Había recuperado los valores de Hb y glóbulos blancos pero el recuento plaquetario era de 99.000/mm3. Se decidió comenzar con metilprednisolona 1g/día por 3 días. Al 3° día el paciente no presentó fiebre y las plaquetas habían ascendido a 135.000/mm3 con GOT: 78U/ml; GPT: 115 U/ml; FAL: 627 U/ml; ferritina: 907 ng/ml. Fue extubado pero requirió reintubación por nuevo sangrado pulmonar con caída de los valores de PaO2. Una nueva PCR para CMV fue positiva. Reinició ganciclovir y metilprednisolona en altas dosis. Requirió diálisis por oligoanuria. Una nueva PAMO mostró médula ósea normal. Con mejoría clínica, se inició destete del respirador. Falleció a los 36 días de internación, luego de una intervención quirúrgica de urgencia por isquemia enterocolónica con hemocultivos positivos a Clostridium perfringens.
Discusión
El SHR es una entidad aún no familiar para el médico intensivista. Los cuatro pacientes presentados en esta comunicación no fueron estudiados ni tratados en forma uniforme. El diagnóstico fue sospechado luego de descartar otras causas más frecuentes y conocidas. Todos ellos presentaron depleción drástica de las tres series sanguíneas. Debido a que la anemia suele ser frecuente en los pacientes internados en UTI, el hallazgo de leucopenia y plaquetopenia suelen ser los datos más llamativos. La fiebre fue el signo presente en todos los pacientes y todos ellos presentaron hemofagocitosis en la primera punción de médula ósea, hallazgo que no es universal. Infecciones bacterianas fueron los desencadenantes del síndrome en 2 pacientes, y una infección viral en otro que había recibido un transplante renal. En el cuarto paciente se consideró el SHR secundario a una infección viral no comprobada.
Gran parte de las internaciones en UTI se deben a cuadros infecciosos, desencadenantes frecuentes del SHR3, 4. El SHR se caracteriza por la presencia de citopenias en al menos dos líneas celulares en el examen de la sangre venosa periférica5. Stéphan y col6 hallaron al SHR como causa de trombocitopenia en más del 50% (12/20) de los pacientes inmunocompetentes ventilados mecánicamente con sepsis o shock séptico. Francois y col7 diagnosticaron 32 casos de SHR en 599 pacientes críticos internados con sepsis y trombocitopenia. En un estudio retrospectivo, Strauss y col.8 hallaron evidencia de hemofagocitosis en la médula ósea en 69 de 107 (64.5%) autopsias de pacientes fallecidos en UTI. Otros signos y síntomas son la fiebre, alteraciones de la función hepática y a menudo coagulopatía, hepatoesplenomegalia, adenopatías y rash cutáneo. Sin embargo, el rash, la hepatoesplenomegalia y las adenomegalias no parecen frecuentes en el SHR secundario a bacterias2. El descenso del fibrinógeno, la hipertrigliceridemia, niveles de ferritina elevados y eritrosedimentación baja son otros hallazgos típicos. Las histiocitosis han sido clasificadas por el grupo de trabajo de la Sociedad Histiocitaria (www.histio.org/society) en 3 grupos: 1) desórdenes relacionados a células dendríticas, 2) desórdenes relacionados a macrófagos y 3) desórdenes malignos 9. El SHR pertenece a la segunda categoría, conjuntamente con el Síndome de activación macrofágica (SAM) y la linfohistiocitosis hemofagocítica (LHH) familiar. El término SAM acuñado por Hadchouel y col en 198510 ha sido usado casi exclusivamente para describir una condición similar al SHR en asociación con enfermedades reumáticas, especialmente la artritis reumatoidea juvenil. Los criterios diagnósticos utilizados son los establecidos en el protocolo LHH 2004 (http/www.histio.org/society/protocols/trials-protocols.shtml) el que exige 5 de 8 criterios clínicos, de laboratorio e histopatológicos. La fiebre y la esplenomegalia son los criterios clínicos. A las citopenias, se agregan la hipertrigliceridemia > 265 mg/dl e hipofibrinogenemia < 1.5 g/l como criterios de laboratorio. El criterio diagnóstico histopatológico es la confirmación de hemofagocitosis en médula ósea, bazo o ganglios linfáticos. Arbitrariamente se ha adoptado el criterio de Wong y Chang11 que exige para el diagnóstico que > 2% de todas las células nucleadas de la médula ósea sean histiocitos con hemofagocitosis. La hemocitofagia medular tiene un rédito del 75%. La hemofagocitosis es más frecuentemente hallada en el hígado y bazo, pero estos sitios son difíciles si no imposibles de biopsiar en los pacientes críticos o con coagulopatía. Por esta razón, se sugiere repetir las muestras de médula ante resultados negativos con un cuadro compatible12.
La sepsis grave se asocia con fallo multiorgánico (FMO). La relación entre SHR y FMO no es clara. Reiner13 reportó 4 casos de FMO entre 23 casos de SHR. Gauvin14 describió 2 casos pediátricos de sepsis con FMO y SHR, sugiriendo que el SHR puede ser parte integral del FMO. Ambas entidades comparten desencadenantes infecciosos entre los que se encuentran virus, bacterias, hongos y parásitos. En ambas está presente un nivel aumentado de citoquinas. Si bien esto no es prueba de su vinculación, sugiere que están asociadas a un estado proinflamatorio. Los hallazgos clínicos durante la fase aguda del SHR pueden ser explicados como consecuencia de la acción de citoquinas originadas presumiblemente en macrófagos y células T activadas. La anormalidad inmunológica más frecuente es la alteración global de la función citotóxica. La función de las células NK está marcadamente disminuida o ausente y la actividad citotóxica de los CD8+ también es defectuosa15. Muchos marcadores de la activación de los macrófagos (ferritina, ß2 microglobulina, enolasa neuronoespecífica) y citoquinas (interferón gamma, factor de necrosis tumoral alfa e interleuquinas 12 y 8) están aumentadas durante el SHR. Frecuentemente se detectan altas concentraciones de la cadena alfa del receptor soluble IL-2 (sCD 25), valores superiores a 2.400 µ/ml es un nuevo criterio diagnóstico y se asocia con peor pronóstico. Inicialmente, la sepsis se caracteriza por un incremento de los mediadores inflamatorios. La presencia de bacterias activa a los macrófagos, células dendríticas y neutrófilos. Estos tres elementos celulares activan a los linfocitos T CD4 a través de mediadores. Las células NK y los linfocitos T citotóxicos podrían jugar un papel en los pacientes con sepsis grave y shock séptico. Su actividad puede medirse directamente (gránulos A y B) e indirectamente a través de citoquinas (IL-12p40) en los pacientes sépticos y su aumento estaría asociado a mayor y a disfunción multiorgánica mortalidad16.
El tratamiento de la sepsis y del SHR difieren completamente. La respuesta al tratamiento es en general peor en los adultos con SHR por razones no conocidas totalmente. Los objetivos del tratamiento son suprimir la hiperinflamación responsable de los síntomas y destruir a las células infectadas presentadoras de antígeno. Según el protocolo de la Sociedad Histiocitaria revisado en el año 2004 (LHH2004), la terapeútica inicial consiste en corticoesteroides (CE), preferentemente dexametasona que cruza la barrera hematoencefálica mejor que la prednisolona, ciclosporina A (CSA) y etopósido. Los CE son citotóxicos para los linfocitos e inhiben la expresión de citoquinas, la CSA impide la activación de los linfocitos, el etopósido tiene alta actividad en las enfermedades histiocitarias y es particularmente útil en el SHR por virus Epstein Barr. En pacientes con síntomas menos graves, la asociación de CE e inmunoglobulinas puede ser suficiente17. Dos grandes series han mostrado resultados promisorios con el uso de gammaglobulina endovenosa (GGEV), especialmente en el SHR secundario a infecciones18. Debe enfatizarse que el tratamiento dirigido solamente contra el patógeno no es suficiente, con la posible excepción de la leishmaniasis, en la cual el tratamiento con anfotericina B suele ser curativo. La administración de GGEV ha tenido poco impacto en pacientes sépticos no seleccionados. Tres de nuestros pacientes recibieron GGEV y en uno de ellos se asoció con CE, con recuperación de las citopenias; pese a ello todos los pacientes fallecieron. La hiperferritinemia y la hemofagocitosis probablemente definan un subgrupo de pacientes sépticos con mejor respuesta a la GGEV17. El valor sugerido de ferritina para el diagnóstico es >10.000 µg/l o >500 µg/l según los nuevos criterios diagnósticos incorporados en el protocolo LHH. En conclusión, el SHF y la sepsis comparten aspectos clínicos y mecanismos fisiopatológicos, lo que hace que el SHF sea probablemente una entidad subdiagnosticada en las unidades de Terapia Intensiva. Proponemos que, ante pacientes gravemente enfermos con depleción de algunas de las series celulares sanguíneas, el hallazgo de hiperferritinemia oriente a la realización de una punción de médula ósea en busca de hemofagocitosis a fin de iniciar rápidamente el tratamiento específico.
Bibliografía
1. Chandra P, Chaudery SA, Rosner F et al. Transient histiocytosis with striking phagocytosis of platelets, leukocytes and erythrocytes. Arch Intern Med 1975; 135: 989-91.
2. Risdall RJ, McKenna RW, Nesbit MF, et al. Virus-associated hemophagocytic syndrome: A benign histyocitic proliferation distinct from malignant histiocytosis. Cancer 1979; 44: 993-02.
3. Risdall RJ. Brunning RD, Hernandez JI, Gordon DH. Bacteria -associated hemophagocytic syndrome. Cancer 1984; 54: 2968-72.
4. Reiner AP, Spivak JL. Hematophagic histiocytosis: a report of 23 new patients and a review of the literature. Medicine (Baltimore) 1988; 67: 369-88.
5. Henter JI, Elinder G, Ost A, et al. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. Semin Oncol 1991; 18: 29-33.
6. Stéphan F, Thioliere B, Verdy E and Tulliez M. Role of Hemophagocitic histiocytosis in the etiology of trombocytopenia in patients with sepsis syndrome or septic shock. Clin Infect Dis 1997; 25: 1159-64.
7. Francois B, Trimoreau F, Vignon P, Prahoran V, Gastinne H. Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating factor. Am J Med 1997; 103: 114-20.
8. Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients. A postmortem clinicopathologic analysis. Crit Care Med 2004; 32:1316-21.
9. Favara BH, Feller AC, Pauli M, et al. Comtemporary classification of histiocytic disorders. The WHO Committee on Histiocytic/Reticulum cell proliferations. Reclassification Working Group of the Histiocytic Society. Med Pediatr Oncol 1997; 29: 157-66.
10. Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic and neurologic disease in juvenile rheumatoid arthritis. Possible relationship with drugs or infection. J Pediatr 1985; 106: 561-6.
11. Wong KF, Chan JKC. Reactive hemophagocytic syndrome: a clinicopathologic study of 40 patients in an oriental population. Am J Med 1992; 93: 177-80.
12. Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol 2002; 14: 548-55.
13. Reiner AP, Spivak JL. Hematophagic histiocytosis. Medicine 1988; 135: 989-91.
14. Gauvin F, Toledano B, Champagne J, Lacroix J. Reactive hemophagocytic syndrome presenting as a component of multiple organ dysfunction syndrome. Crit Care Med 2000; 28: 3341-45.
15. Egeler PM, Shapiro R, Loechelt B, et al. Characteristic inmune abnormalities in hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 1996; 18: 340-5.
16. Zeerleder S, Hack CE, Caliezi C, et al. Activated cytotoxic T cells and NK cells in severe sepsis and septic shock and their role in multiple organ dysfunction. Clin Immunol 2005; 116: 158-65.
17. Larroche C, Bruneel F, Andre MH, et al. Intravenously administered gamma-globulins in reactive hemophago-cytic syndrome. Multicenter study to assess their importance by the immunoglobulins group of experts of CEDIT of the AP-HP. Ann Med Interne (Pans) 2000; 151: 533-9.
18. Emmenegger U, Schaer DJ, Larroche C, Neftel KA. Haemophagocytic syndromes in adults: current concepts and challenges ahead. Swiss Med Wky 2005; 135: 299-314. [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ]
Recibido: 3-05-2006
Aceptado: 28-09-2006

