










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.67 n.2 Buenos Aires mar./abr. 2007
Modelos animales de lesión y reparación del cerebro en desarrollo
Eduardo Cuestas1, Alfredo Cáceres2, Santiago Palacio3
1 Servicio de Pediatría y Neonatología, Hospital Privado;
2 Instituto de Investigaciones Médicas Mercedes y Martín Ferreyra, INIMEC-CONICET,
3 Servicio de Neurología, Hospital Privado, 2da. Cátedra de Neurología, Facultad de Ciencias Médicas, Universidad Nacional de Córdoba
Dirección postal: Dr. Eduardo Cuestas, Servicio de Pediatría y Neonatología. Hospital Privado, Naciones Unidas 346, 5016 Córdoba, Argentina. Fax: (54) 0351-4688286
e-mail: ecuestas@hospitalprivadosa.com.ar
Resumen
Gran parte de la morbilidad y mortalidad neonatal están determinadas por la lesión del cerebro en desarrollo. Un considerable número de los niños afectados presentarán secuelas neurológicas a largo plazo. A pesar de la importancia médica y social que presenta el problema, los avances alcanzados por la medicina neonatal no cuentan aún con una terapéutica eficaz para prevenir o aminorar las consecuencias de la lesión del cerebro en desarrollo. En la siguiente revisión nos proponemos actualizar las investigaciones más recientes en relación a los mecanismos de lesión y reparación del cerebro en desarrollo, basados en modelos animales que ilustran sobre los mecanismos plásticos de adaptación neuronal y funcional; el fin es un mejor conocimiento de los citados procesos que ayude al clínico en la práctica cotidiana de la neonatología.
Palabras clave: Lesión; Reparación; Cerebro en desarrollo
Abstract
Animal models of injury and repair in developing brain. Brain injury is a major contributor to neonatal morbidity and mortality, a considerable group of these children will develop long term neurological sequels. Despite the great clinical and social significance and the advances in neonatal medicine, no therapy yet does exist that prevent or decrease detrimental effects in cases of neonatal brain injury. Our objective was to review recent research in relation to the hypothesis for repair mechanism in the developing brain, based in animal models that show developmental compensatory mechanisms that promote neural and functional plasticity. A better understanding of these adaptive mechanisms will help clinicians to apply knowledge derived from animals to human clinical situations.
Key words: Injury; Repair; Developing brain
Los progresos recientes de la medicina neonatal han permitido disminuir la mortalidad y la morbilidad a largo plazo, especialmente en los recién nacidos con edad gestacional menor de 32 semanas1; este fenómeno se debe en parte al conocimiento más detallado de la patología de los neonatos prematuros y al mejor cuidado intensivo de los recién nacidos con bajo peso extremo (< 1250 g). Aunque la incidencia de hemorragia intraventricular y de leucomalacia periventricular han disminuido en este grupo de pacientes, el nacimiento prematuro presenta todavía, a largo plazo, una frecuencia considerable de discapacidad neurológica, conductual y cognitiva.
Varios trabajos de investigación sugieren que existe la capacidad para reparar las lesiones del cerebro en desarrollo. Se han documentado mecanismos compensatorios del desarrollo que pueden promover plasticidad neuronal y funcional, evidenciando la habilidad del cerebro para regenerar y reemplazar en forma potencial células dañadas2.
En el presente escrito revisamos la bibliografía disponible sobre los procesos de lesión del cerebro prematuro y analizamos las hipótesis sobre los mecanismos de reparación de índole molecular y celular en los diferentes estadios evolutivos del cerebro en desarrollo basados en modelos animales. Un mejor conocimiento de estos mecanismos de adaptación moleculares y celulares puede ayudar al clínico a entender, aplicar y resolver situaciones que potencialmente requieran protección del cerebro en desarrollo, durante el diario quehacer de la medicina neonatal.
El desarrollo único del cerebro humano
El proceso de desarrollo de la neocorteza en el ser humano es admirablemente plástico y se desenvuelve con un patrón de progreso constante. Cualquier distinción entre el cerebro humano y el de otros primates y mamíferos inferiores, reside en las características particulares del desarrollo del hombre propiamente dicho, especialmente el del cerebro. Se han identificado al menos cinco elementos que diferencian al desarrollo del cerebro humano del de otras especies de mamíferos, estos son: 1) La conservación del crecimiento neuronal fetal después del nacimiento; 2) la migración de las células hacia el tálamo dorsal; 3) la elevada actividad transcripcional; 4) la forma específica del gen FOXP2, como crítico para el habla y el lenguaje; y 5) la continuación de la maduración del cerebro en la vida adulta. Conociendo estos elementos y basados en observaciones clínicas de patrones de lesión, ciertos grupos de investigadores han intentado crear modelos animales de lesión y reparación del cerebro en desarrollo que provean información útil para ser transferida al ámbito clínico de la medicina perinatal.Observaciones clínicas de los patrones de lesión en el recién nacido humano
Los patrones de lesión después de una injuria hipóxico-isquémica sobre el cerebro en desarrollo dependen de la gravedad del insulto y de la edad en que se produce. Sabemos por estudios seriados basados en imágenes realizados en seres humanos y confirmados por histopatología, que las lesiones evolucionan en días o aun en semanas. Las diferentes regiones del cerebro presentan distintas susceptibilidades a la injuria dependiendo de su estado madurativo. El cerebro inmaduro responde al insulto hipóxico-isquémico cuando éste ocurre temprano en la gestación y el producto nace prematuramente, con la pérdida de una importante cantidad de oligodendrocitos en desarrollo y de las neuronas inferiores de la placa neurogénica; estas últimas células aparecen transitoriamente durante el desarrollo del sistema nervioso y juegan un rol crítico en la formación de las conexiones entre el tálamo y la corteza visual. En el neonato a término, en cambio, la injuria hipóxico-isquémica afecta determinadas neuronas en los núcleos grises de la base y la corteza perirolándica. Otras células nerviosas, fundamentalmente las que expresan la óxido nítrico-sintetasa, son más resistentes a la hipoxia; dichas células, en los núcleos basales, participan además en los procesos de estrés oxidativo y excitotoxicidad que llevan a la muerte a las neuronas vecinas. La inflamación y la apoptosis mediante los receptores de citoquinas, son otros de los mecanismos involucrados de daño y muerte celular (Fig. 1).
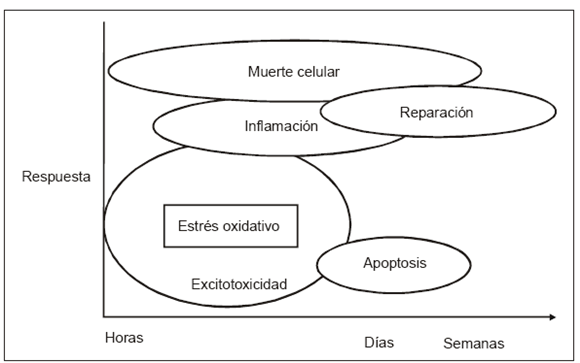
Fig. 1.- Mecanismos de lesión cerebral en el recién nacido.
Las crías de roedores constituyen un modelo animal adecuado para estudiar el cerebro prematuro frente al insulto hipóxico-isquémico
Las variadas discapacidades en el desarrollo neurológico que presentan los prematuros de muy bajo peso al nacer, sugieren que el nacimiento prematuro interrumpe patrones genéticamente programados en la génesis cerebral.
Para desarrollar un modelo clínicamente relevante de los efectos del nacimiento prematuro sobre el cerebro en desarrollo, se debe utilizar un modelo animal que muestre que la injuria infligida experimentalmente resulta en cambios neuropatológicos similares a los observados en los niños prematuros, y que dichos cambios se relacionen con las conductas y alteraciones clínicas presentes en este grupo de pacientes3 . Al igual que el cerebro del prematuro humano al final del segundo trimestre de gestación, la generación neuronal en el equivalente múrido está completada en la mayoría de las regiones cerebrales, junto a una pronunciada proliferación axonal y dendrítica, mientras que la sinaptogénesis está recién comenzando, constituyendo de esta manera un modelo análogo de desarrollo cerebral.
La literatura indica que la privación de oxígeno es la causa más importante de las discapacidades en el desarrollo neurológico que presentan los recién nacidos prematuros4, 5. Aunque la hemorragia intraventricular, la leucomalasia periventricular6 y la ventriculomegalia son las entidades más comúnmente reconocidas y estudiadas dentro de las alteraciones producidas por las anomalías del flujo sanguíneo cerebral, la hipoxemia es particularmente prevalente entre los recién nacidos de muy bajo peso, y es el denominador común de estas anormalidades. Las investigaciones en modelos de hipoxia-isquemia e hipoxia simultáneas7 sobre roedores prematuros han mostrado lesiones focales del cerebro en desarrollo, mientras que la exposición de animales jóvenes solamente a hipoxia resulta en lesión global del cerebro. En crías de ratas y ratones expuestas a períodos considerables de hipoxia se ha observado disminución del peso cerebral, de los volúmenes corticales y del tamaño neuronal, junto a ventriculomegalia. El desarrollo de las espinas dendríticas también está deteriorado en estos animales8. Un reciente análisis molecular de los efectos de la hipoxia crónica sub-letal sobre ratones recién nacidos muestra disrupción en aquellos genes encargados de la sinaptogénesis9. Posteriormente, los animales que sobreviven a la hipoxia crónica en el período neonatal presentan hiperactividad y deterioro en la memoria espacial.
Tomados en conjunto, estos datos permiten inferir que la hipoxia crónica resulta en alteraciones significativas en el desarrollo y la maduración del cerebro en roedores recién nacidos, las que son similares a las que se encuentran en los recién nacidos humanos de muy bajo peso al nacer.
El síndrome de encefalopatía neonatal humana
La encefalopatía neonatal humana se expresa clínicamente como una encefalopatía que evoluciona desde el letargo o la hiperexitabilidad al estupor, en días o semanas. Pero muy a menudo, es una entidad que elude el diagnóstico, especialmente en los prematuros de muy bajo peso, pues la clínica sugerente del síndrome no se presenta a esta edad, o es atribuida muchas veces a la misma inmadurez. La gran frecuencia de presentación de signos y síntomas sutiles conducen comúnmente a un retraso del diagnóstico de parálisis cerebral, de dificultades en el aprendizaje, y de trastornos complejos de la conducta hasta edades muy avanzadas. Fuera de la encefalopatía propiamente dicha, la lesión cerebral puede manifestarse luego como síndrome convulsivo de presentación inusual10 o una encefalopatía isquémica del niño o del joven.
Neurogénesis postnatal y reorganización del cerebro en desarrollo luego de una lesión
Múltiples estudios en crías de ratas y ratones han demostrado que el cerebro puede reorganizar patrones de conexiones para recobrarse o compensar las lesiones durante el desarrollo11; este fenómeno de plasticidad se ha atribuido tanto al incremento de la neurogénesis y de la sinaptogénesis como a la reorganización de circuitos preexistentes12. Se considera plausible que los recién nacidos presenten patrones de reorganización de las conexiones neuronales cuando éstas se encuentran aún en un período de refinamiento; sin embargo, la forma como el cerebro en desarrollo se recupera realmente de la injuria, sea por regeneración celular o por reconstrucción de circuitos preexistentes, es materia de controversia.
Se sabe desde hace algunos años que ciertas regiones del cerebro postnatal y aun adulto, contienen células madre capaces de constituirse en células neurogénicas, pero hace relativamente poco tiempo se ha demostrado este fenómeno en una variedad considerable de mamíferos, incluyendo el hombre13, 14. Aunque las células madre están presentes en todo el cerebro, sólo aquellas ubicadas en la zona sub-ventricular del cerebro anterior y en la capa sub-granular del núcleo dentado parecen ser capaces de neurogénesis in vivo. Estas dos regiones proveen de neuronas al bulbo olfatorio y al propio núcleo dentado respectivamente.
Experimentalmente, las células granulosas generadas de novo en el hipocampo han mostrado integrarse por sí mismas dentro de circuitos preexistentes, tornarse eléctricamente activas y formar conexiones sinápticas15,16.
La neurogénesis postnatal está influenciada tanto por el medio interno como por el externo. La neurogénesis en el hipocampo declina con la edad y es suprimida por el estrés17, 18; en contraste, la proliferación de células progenitoras en el hipocampo es estimulada por los estrógenos y por el ejercicio, mientras la supervivencia de las neuronas recién generadas es promovida por un medio pericelular enriquecido19. Los estímulos ambientales positivos incrementan la sobrevida neuronal; en consecuencia, la intervención temprana protege las neuronas lesionadas, posiblemente debido al aumento de las concentraciones de neurotrofinas en el hipocampo20. Como el enriquecimiento del medio mejora la memoria espacial en los modelos animales, alcanzar una neurogénesis suficiente y un mantenimiento de las condiciones de sobrevida neuronal en el núcleo dentado puede incrementar la habilidad de un animal para adquirir nueva información.
Las células madre de la zona sub-ventricular postnatal y del núcleo dentado también son capaces de responder a una variedad de estímulos nocivos del medio interno. En el ratón adulto, las células madre pueden reconstruir casi la totalidad de la zona sub-ventricular después de un 90% de destrucción de la región21. Estas células progenitoras dan origen a las neuronas piramidales corticales, si es que ha ocurrido una apoptosis generalizada de la corteza cerebral, y también a las neuronas piramidales del hipocampo y del núcleo estriado después de la isquemia experimental en ratas adultas22. Las neuronas recién generadas son dirigidas hacia la región lesionada, sugiriendo que la neuronogénesis es inducida por cambios locales en la expresión genética luego de la injuria23. También se ha descripto regeneración superior al 40% en la capa piramidal CA1 del hipocampo después de isquemia en ratas adultas, pero sólo con posterioridad a la infusión de factor de crecimiento de los fibroblastos (FGF) y de factor de crecimiento epidérmico (EGF) en los ventrículos cerebrales. Como el grado de regeneración se acompaña de una sustancial recuperación de la conducta24, la elucidación de los mecanismos que median los eventos de regeneración neuronal son esenciales para desarrollar estrategias en el tratamiento de la lesión cerebral. Recientemente se ha sugerido que la recuperación de la lesión en el cerebro prematuro incluye la reactivación de la glía radial en las capas germinales, ya que en curso de los días posteriores al insulto isquémico se observa un incremento de la proliferación celular en la zona sub-ventricular y en el núcleo dentado; estas células que comienzan a dividirse luego de la injuria hipóxica parecen ser una forma fenotípicamente determinada de glía radial. Incluso los astrocitos son capaces de "rejuvenecer" y reconvertirse en células neuronogénicas. Recordemos que los astrocitos se originaron a su vez en las células madre de la glía radial, que constituyen los progenitores embrionarios mitóticamente activos que normalmente participan en el desarrollo del neuroepitelio emitiendo prolongaciones basales y apicales entre las capas ventricular y la pia. Dichas células serían las encargadas de generar las neuronas corticales durante la embriogénesis, mediante los procesos de mitosis simétrica y asimétrica, realizando largas migraciones desde su origen hasta su destino final o blanco en las diferentes capas de la corteza. Después que la corticogénesis termina, la glía radial gradualmente retrae las prolongaciones ventriculares dentro de los astrocitos multipolares, finalizando el proceso de neuronogénesis y, en este caso particular, de recuperación luego de un insulto hipóxico-isquémico25.
Intervención y neuroprotección farmacológica
Los modelos de intervención en animales han demostrado que el cerebro inmaduro responde de manera diferente al cerebro adulto frente a la hipoxia-isquemia. Las terapéuticas desarrolladas para disminuir la lesión cerebral el adultos pueden empeorar los resultados en los recién nacidos, posiblemente por acentuación de la apoptosis. Las drogas que bloquean los receptores de n-metil-d-aspartato o que potencian los receptores de ácido g-aminobutírico disparan amplios mecanismos de apoptosis en los roedores prematuros. Por esto, las drogas que en sentido inverso activan estos receptores, como el midazolan, el óxido nítrico y el isoflourano, producen una mejoría en la capacidad de aprendizaje cuando se aplican a ratas de 7 días sometidas a hipoxemia. Otros principios activos como el alopurinol, la desferoxamina y la 3-imibiotina interrumpen la injuria causada por los radicales libres y han demostrado ser beneficiosos en varios modelos animales4. Los efectos neuroprotectores de la eritropoyetina han recibido recientemente mucha atención26.
Las células de la guía glial y la recuperación de la lesión del cerebro en desarrollo
Recapitulando, entendemos que se ha demostrado experimentalmente que la recuperación de la lesión en el cerebro del prematuro implica la reactivación de la glía radial en las capas germinales27. Por lo tanto, unos días después de la injuria hipóxica aguda o crónica, se presenta un aumento de la proliferación celular tanto en la zona sub-ventricular como en el núcleo dentado, y las células reactivadas comienzan a dividirse después de la hipoxia; dichas células parecen ser también fenotípicamente una forma de glía radial. Esta abundancia de glía radial tal vez sea secundaria al incremento de la proliferación de dichas células después de la lesión, o a una reversión de su involución en astrocitos; por lo tanto, luego de una lesión cerebral, los astrocitos son capaces de rejuvenecer revirtiendo su proceso madurativo nuevamente hacia glía radial la cual, a su turno, permitirá generar nuevas neuronas. Sabemos ahora que el cerebro de los mamíferos aun después de la embriogénesis, conserva células madre neurogénicas en la zona sub-ventricular, especialmente en la pared lateral de los ventrículos. Estas células generan nuevas neuronas que migran a través de la corriente migratoria rostral hacia el bulbo olfatorio, donde se diferencian en interneuronas granulares y periglomerulares. Aunque dichas células son activamente neurogénicas por división asimétrica, en esta fase se presentan morfológica y ultraestructuralmente como astrocitos, teniendo la peculiaridad de expresar proteínas acídicas fibrilares gliales (GFAP). Estos hallazgos desafían el punto de vista clásico de que los astrocitos se diferencian finalmente de la glía y pertenecen a un linaje separado de las neuronas. En la actualidad, la hipótesis más aceptada es que las células madre de la zona sub-ventricular derivan de la glía radial. Esta se caracteriza por presentar prolongados procesos basales tipo RC2 positivos, que se extienden desde sus cuerpos celulares en la zona ventricular hacia el parénquima a través de la superficie cerebral, presentando también los rasgos anatómicos de las células del linaje astroglial, incluyendo prolongaciones envolventes sobre los vasos sanguíneos, filamentos intermedios y gránulos de glucógeno. Como al final de la histogénesis algunas células de la glía radial se diferencian en astrocitos parenquimatosos, clásicamente ésta era vista como una forma celular inmadura, cuya función era guiar a las neuronas en su migración e intervenir en la determinación neuronal para el desarrollo cerebral. Sin embargo, la observación de que en las aves canoras la glía radial persiste hasta la vida adulta, junto a los hallazgos realizados en cerebros de roedores en desarrollo, demuestran que la glía radial da origen a neuronas, células ependimales, oligodendrocitos y astrocitos propiamente dichos, pudiendo además revertir nuevamente dicho ciclo, hacia células del sistema macroglial.
La glía radial expresa genéticamente receptores para el factor de crecimiento de los fibroblastos (FGF), y varios estudios in vitro sugieren al factor de crecimiento de fibroblastos 2 (FGF2) como inductor de la proliferación y expansión de esos progenitores. El FGF2 podría también tener importancia en la regeneración del cerebro postnatal, ya que su concentración está aumentada en la fase de recuperación después de la hipoxia, así como la expresión de los receptores para FGF1 en la zona subventricular28. Además hay que tener en cuenta los factores genéticos y ambientales que tienen influencia sobre la neurogénesis, tales como la secreción de factores de crecimiento y la "intervención temprana" por enriquecimiento del medio, que ciertamente pueden afectar una variedad de procesos de crecimiento que incluyen la sinaptogénesis y la formación de fibras nerviosas. Es entonces esencial poder discriminar dentro de un gran número de eventos, aquellos que son críticos en forma directa para la recuperación funcional. Son candidatos potenciales para esto los genes que regulan los factores de crecimiento, sus receptores, los mecanismos de transducción intracelular, los mecanismos que regulan la apoptosis, y los factores de transcripción que intervienen dentro de las células madre para generar determinadas líneas celulares. Los modelos adaptativos transgénicos en ratones que expresen positiva o negativamente estas moléculas y sean expuestos a hipoxia sub-letal, podrían representar adecuadamente los mecanismos de desarrollo del cerebro prematuro; además, estos modelos permitirían recapitular el tipo de daño que se presenta en los recién nacidos prematuros, permitiendo probar mecanismos de recuperación y potenciales tratamientos.
Las células gliales primitivas como las de la glía radial pueden ser capaces de generar nuevas células para reparar el cerebro bajo condiciones en las que las células maduras están muriendo, permitiendo suponer que el cerebro del recién nacido prematuro puede ser capaz de desarrollar programas de regeneración luego de la lesión hipóxico-isquémica (Fig. 2). Restan aún por investigar los mecanismos que promueven la proliferación de esas células, su diferenciación en neuronas y células gliales, y su posterior integración en circuitos nerviosos funcionales.
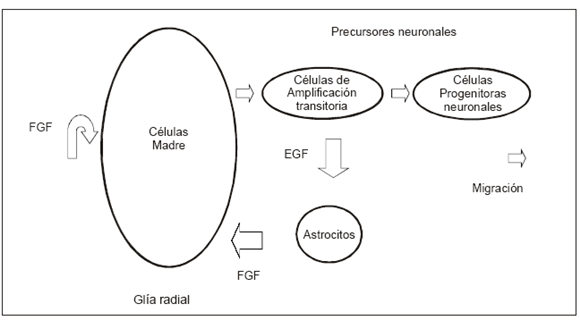
Fig. 2.- Mecanismos de regeneración neuronal del cerebro en desarrollo.FCF: factor de crecimiento de los fibroblastos, EGF: factor de crecimiento epidérmico
Bibliografía
1. Hack M, Wrigth LL, Shankaran S, et al. Very-low-birthweight outcomes of the National Institute of Child Health and Human Development Neonatal Network, November 1989 to October 1990. Am J Obstet Gynecol 1995; 172: 457-64.
2. Volpe JJ. Perinatal brain injury: from pathogenesis to neuroprotection. Ment Retard Dev Disabil Res Rev 2001; 7: 56-64.
3. Dobbing J. Undernutrition and the developing brain: the use of animal models to elucidate the human problems. Psychiat Neurol Neurochir 1971; 74: 433-42.
4. Ferriero DM. Neonatal brain injury. N Engl J Med 2004; 351: 1985-95.
5. Vexler ZS, Ferriero DM. Molecular and biochemical mechanisms of perinatal brain injury. Semin Neonatol 2001; 6: 99-108.
6. Back SA, Volpe JJ. Cellular and molecular pathogenesis of periventricular white matter injury. Ment Retard Dev Disabil Res Rev 1997; 3: 96-107.
7. Nyakas C, Buwalda B, Luiten P. Hypoxia and brain development. Prog Neurobiol 1996; 49: 1-51.
8. Szebenyi G, Bollati F, Bisbal M, et al. Activity-driven dendritic remodeling requires microtubule-associated protein 1A. Curr Biol 2005;15: 1820-6.
9. Curristin SM, Cao A, Steward WB, et al. Disrupted synaptic development in the hypoxic newborn brain. Proc Natl Acad Sci USA 2002; 99: 1529-34.
10. Cuestas E. Is routine EEG helpful in the management of complex febrile seizures? Arch Dis Child. 2004; 89: 290.
11. Kitagawa K, Motsumoto M, Hari M. Protective and regenerative responses endogenously induced in the ischemic brain. Can J Physiol Pharmacol 2001; 79: 262-5.
12. Alvarez-Buylla A, Herrera DG, Wichterle H. The subventricular zone: source of neural precursors for brain repair. Prog Brain Res 2000; 127: 1-11.
13. Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogesis in rats. J Comp Neurol 1965; 124: 319-35.
14. Eriksson PS, Perfilieva E, Bjork-Eriksson T, et al. Neurogenesis in the adult human hippocampus (comment). Nat Med 1998; 4: 1313-17.
15. Song HJ, Stevens CF, Gage FH. Neuronal stem cells from adult hippocampus develop essential properties of functional CNS neurons. Nat Neurosci 2002; 5: 438-45.
16. Cáceres A, Quiroga S. Regulation of membrane expansion at nerve growth cone. J Cell Sci 2003; 116: 1209-17.
17. Gould E, Tanpat P, McEwen BS, et al. Proliferation of granule cell precursors in the dentate gyrus of adults monkeys is disminished by stress. Proc Natl Acad Sci USA 1998; 95: 3168-71.
18. Kemperman G, Kuhn HG, Gage FH. Experience-induced neurogenesis in the senescent dentate gyrus. J Neurosci 1998; 18: 3206-12.
19. Tanapat P, Hastings NB, Reeves AJ, et al. Estrogen stimulates the proliferation of granule cell precursors in the dentate gyrus of adult females rats. Neuroscience Abstracts 1998; 24: 796-8.
20. Young D, Lawlor PA, Leone P, et al. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat Med 1999; 5: 448-53.
21. Doetsch F, García-Verdugo JM, Alvarez-Builla A. Rege-neration of germinal layer in the adult mammalian brain. Proc Natl Acad Sci USA 1999; 11919-24.
22. Magavi SS, Macklis JD. Induction of neuronal type-specific neurogenesis in the cerebral cortex of adult mice: manipulation of neural precursors in situ. Dev Brain Res 2002; 134: 57-76.
23. Wang Y, Sheen VL, Macklis JD. Cortical interneurons upregulate neurotrofins in vivo in response to targered apoptotic degeneration of neighboring pyramidal neurons. Exp Neurol 1998; 154: 389-402.
24. Nakatomi H, Kuriu T, Okabe S, et al. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural precursors. Cell 2002; 110: 429-41.
25. Doetsch F, Caille I, Lim DA, et al. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999; 97: 703-16.
26. Solá A, Wen T-C, Hamrick SEG, et al. Potential protec-tion and repair following injury to the developing brain: a role for erythropoietin? Ped Res 2005; 57: 110R-17R.
27. Ganat Y, Soni S, Chacon M, et al. Chronic hypoxia up-regulates fibroblast growth factors ligands in the perinatal brain and induces fibroblast growth factor-responsive radial glial cell in the sub-ependymal zone. Neuroscience 2002; 112: 977-91.
28. Vaccarino FM, Schartz ML, Hartigan D, et al. Effect of basic fibroblast growth factor on the genesis of excitatory and inhibitory neurons in primary cultures of cells from the mammalian telencephalon. Cereb Cortex 1995; 1: 64-78. [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ]

