










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.67 n.3 Buenos Aires mayo/jun. 2007
Expresión inmunohistoquímica e inestabilidad microsatelital en el síndrome de Lynch
Carlos A. Vaccaro1, Jorge E. Carrozzo1, Esteban Mocetti2, Mariana Berho3, Paula Valdemoros2, Eduardo Mullen2, Myriam Oviedo4, María A. Redal5
1 Sección de Coloproctología y
2 Servicio de Anatomía Patológica, Hospital Italiano de Buenos Aires;
3 Departamento de Anatomía Patológica y
4 Departamento de Cirugía Colorrectal, Cleveland Clinic Florida Weston, EE.UU.;
5 Instituto de Ciencias Básicas y Medicina Experimental, Hospital Italiano de Buenos Aires
Dirección postal: Dr. Carlos A. Vaccaro, Sección de Coloproctología, Hospital Italiano, Gascón 450, 1181 Buenos Aires, Argentina. Fax: (54-11) 4958-2200 e-mail: carlos.vaccaro@hospitalitaliano.org.ar
Resumen
Las mutaciones de los genes MLH1 y MSH2 son frecuentemente implicadas en el síndrome de Lynch. La expresión inmunohistoquímica (IHQ) es una forma simple de selección para pruebas moleculares. Se analizó la IHQ de MLH1 y MSH2 en pacientes con síndrome de Lynch (16 casos) y pacientes menores de 50 años sin antecedentes familiares (25 casos). Se estudiaron 41 tumores de un grupo de pacientes (64% mujeres) de edad promedio 40.7 años (rango: 16-75). Se obtuvieron resultados concluyentes en 40 casos (97.6%). Dieciocho casos (45%) presentaron falta de expresión (MLH1 negativa: 11 casos; MSH2 negativa: 6 casos; MLH1 negativa y MSH2 negativa: 1 caso), con una incidencia significativamente mayor en pacientes con síndrome de Lynch (68.7% vs. 28%, p=0.01). Entre los casos esporádicos, 5 casos (20%) mostraron falta de expresión MLH1 y 2 casos (8%) con falta de expresión MSH2. La falta de expresión IHQ presentó una fuerte asociación con inestabilidad microsatelital alta (IMS): expresión normal: 5.9%, expresión negativa: 92.3%, P<0.0001. Los índices de sensibilidad y especificidad de la IHQ para detectar IMS fueron de 92.3% y 94.1% respectivamente. Los patrones de IHQ y de IMS no se relacionaron a ninguna característica histopatológica. En conclusión, el análisis inmunohistoquímico de las proteínas MLH1 y MSH2 fue altamente sensible y específico para detectar IMS y permitió identificar en un 45% de los casos la proteína alterada. El índice de falta de expresión IHQ entre los casos esporádicos diagnosticados antes de los 50 años justifica su implementación sistemática en este grupo de pacientes.
Palabras clave: Síndrome de Lynch; Cáncer colorrectal; Inmunohistoquímica; Inestabilidad microsatelital
Abstract
Immunohistochemical expression and microsatellite instability in Lynch syndrome. Mutation of the mismatch repair genes MLH1 and MSH2 account for the majority of the genetic abnormalities in Lynch syndrome. Immunohistochemical detection of their protein products is becoming an increasingly common method to detect these mutations. The aim of this study was to compare the expression of MLH1 and MSH2 by immunohistochemistry and its relationship with a group of clinical and histological variables in patients with known Lynch syndrome (n=16) and in cohort of young patients (less than 50 years) who did not meet Amsterdam criteria (n=25). The mean age was 40.7 and 64% were women. Conclusive results were obtained in 40 cases (97.6%). Eighteen cases (45%) showed abnormal expression of either MLH1 (11 cases) or MSH2 (6 cases) and both stains (1 case). Alteration of the normal staining pattern was seen more commonly in patients with Lynch syndrome than in the sporadic group (68.7% vs 28%, p=0.01). A significant correlation was obtained between abnormal protein expression and microsatellite instability (MSI): normal expression: 5.9%, lack of expression: 92.3%, p<0.0001. The sensitivity and specificity of the immunohistochemical to predict MSI were 92.3% and 94.1% respectively. Immunohistochemistry and MSI results did not correlate with any histopathological parameter. In conclusion, in our experience abnormal staining of MLH and MSH correlates strongly with the presence of MSI. In addition it appears that in our population a significant proportion of young patients (< 50 years old) demonstrate alterations in the mismatch repair gene products suggesting an important role of these molecules in tumorigenesis.
Key words: Lynch syndrome; Colorectal cancer; Immunohistochemical expression; Microsatellite instability
El cáncer colorrectal (CCR) es la tercera causa de muerte por cáncer en Occidente1. De acuerdo a los datos del Globocan, en Argentina se diagnostican 10.300 casos nuevos por año y se producen 5.700 muertes. Entre el 3% y el 8% de los CCR se presentan como formas hereditarias, siendo el síndrome de Lynch (cáncer colorrectal hereditario no polipósico) la más frecuente de ellas. Las principales características de esta entidad son su presentación a edades tempranas (promedio 45 años); la afectación predominante del colon derecho (en el 70% de los casos); la alta incidencia de tumores colorrectales sincrónicos (10%) y metacrónicos (30 a 50%) y la asociación con tumores extracolorrectales (especialmente de adenocarcinomas de endometrio, intestino delgado, ovario, estómago y vías urinarias)2. El diagnóstico clínico se sospecha por los antecedentes familiares y se confirma a través de la identificación de mutaciones germinales en los genes reparadores de errores de apareamiento de bases nucleotídicas (fundamentalmente MLH1 y MSH2). Estas mutaciones determinan un estado de inestabilidad genómica que se conoce como inestabilidad microsatelital (IMS)3, alteración que está presente en más del 85% de los tumores del síndrome de Lynch y en el 10% al 15% de los tumores esporádicos. En este último grupo, si bien la alteración primaria suele ser la metilación del gen MLH1, también han sido identificadas mutaciones germinales que son interpretadas como mutaciones de novo.
Tanto la identificación de mutaciones como la determinación de la IMS implican tecnología molecular que resulta altamente costosa y de disponibilidad limitada, especialmente en países en desarrollo. El estudio de la expresión inmunohistoquímica (IHQ) de las proteínas más comúnmente afectadas (MLH1 y MSH2) se presenta como una alternativa muy ventajosa.
El objetivo del presente trabajo es analizar por inmunohistoquímica la expresión proteica MLH1 y MSH2 y establecer su relación con el grado de IMS y el patrón histológico, en pacientes con síndrome de Lynch y con CCR esporádico diagnosticado antes de los 50 años.
Materiales y métodos
Población: se analizaron las piezas de 41 pacientes (edad promedio: 40.7 años; rango 16-75; sexo femenino 65.9%) operados en el Hospital Italiano de Buenos Aires entre enero del 2000 y diciembre del 2005. Dieciséis casos fueron definidos como síndrome de Lynch en base a los Criterios de Amsterdam II4, 5 (Tabla 1). Veinticinco pacientes sin antecedentes familiares y con diagnóstico de CCR antes de los 50 años fueron definidos como esporádicos.
TABLA 1.- Criterios de Amsterdam II para el síndrome de Lynch

Estudio histológico: el grado histológico se categorizó en bien diferenciado (G1), moderadamente diferenciado (G2) y poco diferenciado (G3). Cuando la mucosecreción superó el 50% de la superficie tumoral se lo consideró tumor mucosecretor.
La presencia de linfocitos intratumorales (LIT) fue medida en 5 campos de gran aumento (40X) en el frente de invasión tumoral. Se clasificó en 0 (ausencia de linfocitos), 1 (menor de 2 linfocitos), 2 (entre 2 y 5 linfocitos) y 3 (mayor de 5 linfocitos).
La inflamación tipo Crohn fue definida como la acumulación de linfocitos con o sin formación de centro germinal en el frente de infiltración tumoral.
Expresión Inmunohistoquímica (IHQ): para la inmunomarcación se utilizaron cortes histológicos individuales de 5 µm montados en portaobjetos adherentes con carga positiva. Luego se realizaron técnicas de inmunomarcación con recuperación antigénica (utilizando buffer fórmico 1%/gelatina 2% con pH 2.5%, buffer alcalino AR10, o buffer EDTA 1mM), mediante 4 ciclos de 5 minutos en calor húmedo en microondas a máxima potencia. Se realizó luego incubación con Power Block - Universal Blocking Reagent diluido 1:10 (Biogenex). Posteriormente se efectuó incubación con los anticuerpos primarios monoclonales de ratón anti hMLHI humano (BD Pharmingen Cat. 550838), anti hMSH2 humano (Oncogene, Cat. NA27). Se utilizó como control interno positivo de expresión proteica, la marcación nuclear del tejido normal presente en cada taco evaluado (células epiteliales de mucosa normal, fibroblastos, endotelio, células inflamatorias del estroma o musculares lisas). Se utilizaron controles positivos externos de expresión de MLH1 y MSH2 (epitelio de mucosa colónica normal) y controles externos negativos con déficit de expresión de cada una de las proteínas. Se realizaron además controles internos. El área completa de las secciones histológicas fue evaluada en microscopio óptico en cada caso. Se registró el porcentaje de marcación positiva nuclear en tejido normal y en tejido tumoral comparativamente. Los resultados de las tinciones fueron clasificados como positivos cuando más del 80% de las células presentaban tinción normal, o negativos cuando la tinción no superaba el 10%. Los casos con expresión intermedia fueron definidos como no concluyentes.
Detección de la inestabilidad microsatelital: se estudiaron los microsatélites de ADN de 30 muestras de tumor y mucosa normal. La extracción y purificación de ADN de las muestras se efectuó con el kit QIAamp DNA Mini Kit (QIAgen, Cat. N° 51304). Se realizó la PCR (reacción de cadena de la polimerasa) en un termociclador MJ Research, con primers de secuencia específica para cada uno de los microsatélites en estudios: BAT 25, BAT 26, D2S 123, D5S 346, D17S 250. Se determinó la presencia de producto amplificado mediante una electroforesis en gel de agarosa al 2%, teñido con bromuro de etidio. Para visualizar la inestabilidad genética de los marcadores estudiados, el amplicón fue sometido a una corrida electroforética vertical en gel poliacrilamida, teñido con nitrato de plata.
Se clasificó como inestabilidad microsatelital alta (IMS-Alta) a aquellos casos que presentaban 2 o más microsatélites positivos, baja (IMS-Baja) cuando sólo 1 marcador fue positivo, y estable cuando todos los marcadores fueron negativos. Los estudios de IMS fueron realizados sin previo conocimiento de los resultados de la IHQ.
El estudio se realizó de acuerdo al Consentimiento Médico para participar en Protocolos de Investigación del Comité de Etica de Protocolos de Investigación del Departamento de Docencia e Investigación del Hospital Italiano de Buenos Aires.
Resultados
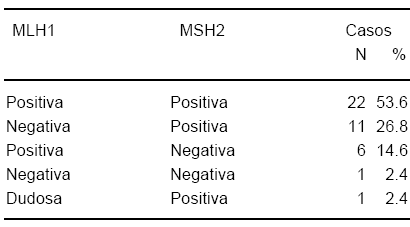
En 40 casos (97.6%) la IHQ pudo calificarse como positiva o negativa mientras que en 1 caso la proteína MLH1 mostró una tinción dudosa (Tabla 2). La falta de expresión de MLH1 y/o MSH2 se observó en 18 casos (45%) con una diferencia significativa entre los grupos (Lynch: 68.7%, Esporádicos: 28%, p=0.01). Entre los pacientes con tumores esporádicos, fueron identificados 5 casos (20%) con falta de expresión MLH1 y 2 casos (8%) con falta de expresión MSH2 (ambos se asociaron a IMS-Alta).
TABLA 2.- Expresión inmunohistoquímica MLH1 y MSH2
De los 30 casos en donde se estudió la inestabilidad de microsatélites, 13 (43.3%) presentaron IMS-Alta, 15 (50%) IMS-Baja y 2 (6.7%) fueron estables. La proporción de tumores con IMS-Alta fue significativamente mayor en los pacientes con síndrome de Lynch que en los jóvenes esporádicos (Tabla 3).
TABLA 3.- Resultados de la determinación de Inestabilidad microsatelital en los diferentes grupos

La expresión IHQ presentó una fuerte asociación con inestabilidad microsatelital alta (expresión normal: 5.9%, expresión negativa: 92.3%, p<0.0001). Los índices de sensibilidad y especificidad de la IHQ para detectar IMS fueron de 92.3% y 94.1% respectivamente.
En cuanto a las variables histológicas y las expresiones de IHQ e IMS los respectivos patrones no se relacionaron a ninguna característica histopatológica, si bien los casos con falta de expresión IHQ mostraron una tendencia a presentarse con una menor proporción de metástasis ganglionares y en colon derecho (Tabla 4 y 5).
TABLA 4.- Variables histológicas y su relación con la expresión inmunohistoquímica
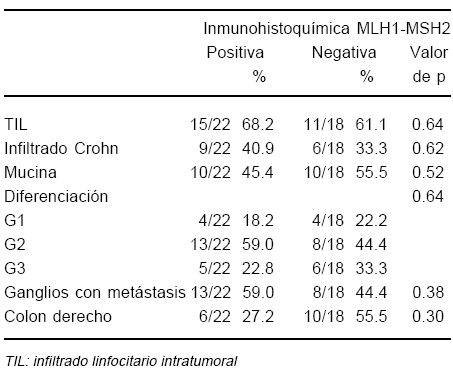
TABLA 5.- Variables histológicas y su relación con estado de microsatélites

Discusión
El síndrome de Lynch suele ser subdiagnosticado debido a la falta de un marcador fenotípico claro6-8. Si bien algunos de estos pacientes presentan antecedentes familiares floridos que permiten hacer el diagnóstico clínico en base a los criterios de Amsterdam4, 5, en aquellos que desconocen sus antecedentes o pertenecen a familias reducidas la única forma de diagnóstico es la identificación de mutaciones germinales9. Existe además un porcentaje de casos, todavía no bien determinado, que representan mutaciones de novo yque se presentan como formas esporádicas pero que son transmisibles en las generaciones subsecuentes10.
La identificación de una mutación implica técnicas altamente costosas y de limitada disponibilidad. Por esta razón, se recomienda utilizar previamente técnicas más sencillas a modo de rastreo, especialmente cuando los criterios de Amsterdam no se cumplimentan11, 12. A tal fin, un panel de expertos desarrolló los denominados criterios de Bethesda13 para seleccionar casos en los que debe realizarse la determinación de IMS. Estos criterios incluyen en primer lugar a todo paciente con diagnóstico de CCR antes de los 50 años (incluyendo las formas esporádicas). De acuerdo a estos lineamientos, en aquellos casos con IMS alta se debe proseguir con el diagnóstico molecular para identificar la mutación germinal.
Alternativamente, se ha propuesto la utilización de la determinación de la expresión IHQ, ya que esta técnica es simple, altamente predictiva de IMS y además permite identificar la proteína alterada. Varios estudios han demostrado que utilizando inmunomarcaciones para MLH1, MSH2, PMS2 y MSH6 la sensibilidad para el diagnóstico de síndrome de Lynch es similar a la utilización de IMS con índices superiores al 90%11, 12, 14.
El presente trabajo representa el primer estudio en nuestro país sobre el perfil inmunohistoquímico y microsatelital de los tumores colorrectales en pacientes con criterios de Amsterdam o con formas esporádicas diagnosticadas antes de los 50 años. En este grupo altamente seleccionado, la inmunohistoquímica fue conclusiva en el 97.6% de los casos y fue altamente sensible y específica para detectar inestabilidad microsatelital. En el caso de los pacientes con diagnóstico de síndrome de Lynch en base a los criterios de Amsterdam, la identificación de la proteína faltante a través de las pruebas de inmunohistoquímica permite limitar los subsecuentes estudios de secuenciación al gen correspondiente. Esta circunstancia se presentó en 11 de 16 casos (68.5%) de la presente serie. El único caso donde hubo una falta de expresión de ambas proteínas, asumimos que la mutación germinal se halla en el gen MSH2 ya que la metilación del mismo no ha sido reportada. En el caso de los pacientes con CCR esporádico, las implicancias son todavía mayores, ya que pueden identificarse mutaciones heredables que de otra manera pueden pasar inadvertidas. En el presente trabajo, identificamos 7 casos (que representan casi un tercio de la serie) donde hubo una falta de expresión normal. Si bien esto puede deberse en parte a la inactivación por hipermetilación del promotor del gen MLH1, algunos pacientes podrían ser portadores de mutaciones germinales (especialmente en los 2 casos en que la falta de expresión correspondió a la proteína MSH2). En consecuencia, el manejo de estos pacientes y sus familiares se ve modificado ya que, independientemente de los antecedentes familiares, las recomendaciones de prevención y seguimiento deben ser similares a aquellos con criterios de Amsterdam. En estos casos, debe proveerse un asesoramiento genético en el marco de un registro multidisciplinario en donde se informe sobre los riesgos oncológicos individuales y familiares, y se discutan las ventajas y desventajas de las pruebas genéticas15, 16, 17. En una reciente evaluación de nuestra experiencia, la totalidad de las personas consideraron que el asesoramiento fue de utilidad y se sintieron satisfechos por la forma en que se les brindó la información. Todas las personas calificaron a la sesión como útil o muy útil y refirieron que se sentían mejor luego de la misma. Sólo 2 de los 34 participantes tuvieron algún grado de duda sobre la confidencialidad de la información18.
En conclusión, el análisis inmunohistoquímico de las proteínas MLH1 y MSH2 fue altamente sensible y específico para detectar inestabilidad microsatelital y permitió identificar en un 45% de los casos, la proteína alterada. El índice de falta de expresión inmunohistoquímica, detectado entre los casos esporádicos diagnosticados antes de los 50 años, tiene importantes implicancias individuales y familiares que justifican su implementación sistemática en este grupo de pacientes.
Bibliografía
1. Jemal A, Murray T, Ward E, et al. Cancer Statistics, 2005. CA Cancer J Clin 2005; 55: 10-30.
2. Umar A, Boland C, Terdiman J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96: 261-8.
3. Soreide K, Janssen EA, Soiland H, et al. Microsatellite instability in colorectal cancer. Br J Surg 2006; 93: 395-406.
4. Vasen HF, Mecklin JP, Khan PM et al. International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34: 424-5.
5. Vasen HF, Watson P, Mecklin JP. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Colla-borative group on HNPCC. Gastroenterology 1999; 116: 1453-6.
6. Church J, Mc Gannon E. Family history of colorectal cancer: how often and how accurately is it recorded? Dis Colon Rectum 2000; 43: 1540-4.
7. Bonadeo Lassalle F, Vaccaro CA, Benati ML, et al. Síndrome de Lynch: implicancias de una patología subdiagnosticada. Rev Argent Cirug 1997; 72: 110-20.
8. Ruo L, Cellini C, La-Calle JP Jr, et al. Limitations of family cancer history assessment at initial surgical consultation. Dis Colon Rectum 2001; 44: 98-103.
9. Percesepe A, Anti M, Roncucci L, et al. The effect of family size on estimates of the frequency of hereditary non-polyposis colorectal cancer. Br J Cancer.1995; 72: 1320-3.
10. Ponz de Leon M. Prevalence of hereditary nonpolyposis colorectal carcinoma (HNPCC). Ann Med.1994; 26: 209-14.
11. Piñol V, Castells A, Andreu M, et al; Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005; 293: 1986-94.
12. O´Reilly S, Johnson KA. Brensinger JD, et al: Hereditary nonpolyposis colorectal cancer. Ann Oncol 1997; 8: 1151-6.
13. Rodríguez-Bigas MA, Boland R, Hamilton SR, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997; 89: 1758-62.
14. Hampel H, Frankel W, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352: 1851-60.
15. Peters JA, Bowles B. Genetic Counseling and hereditary cancer. Cancer supplement. 1997; 80: 576-85.
16. Lerman C, Marshall J, Audrain J, et al. Genetic testing for colon cancer susceptibility: Anticipated reactions of patients and challenges to providers. Int J Cancer 1996; 69: 58-61.
17. Myrhoj T, Bernstein I, Bisgaard ML, et al. The establishment of an HNPCC registers. Anticancer Res.1994; 14: 1647-50.
18. Vaccaro C, Roverano A, Ojea Quintana G, et al. Programa de Cáncer Hereditario: 10 años de experiencia con el síndrome de Lynch. Rev Argent Cirug. En prensa. [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ]
Recibido: 10-05-2006
Aceptado: 18-01-2007

