










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.67 n.4 Buenos Aires jul./ago. 2007
Estudio de 34 pacientes con incidentaloma suprarrenal
Raúl A. Chervin, Karina Danilowicz, Fabián Pitoia, Reynaldo M. Gómez, Oscar D. Bruno
División Endocrinología, Hospital de Clínicas José de San Martín, Facultad de Medicina, Universidad de Buenos Aires
Dirección Postal: Dr. Raúl A. Chervin, División Endocrinología, Hospital de Clínicas, Córdoba 2351, 1120 Buenos Aires, Argentina Fax (54-11) 4805-0631 e-mail: rachervin@intramed.net
Resumen
El incidentaloma suprarrenal, un tumor de dicha glándula descubierto por razones independientes de la sospecha de enfermedad adrenal, constituye un problema clínico frecuente. Aunque en la mayoría de los casos son benignos y no hiperfuncionantes, es importante identificar oportunamente la minoría de lesiones malignas o hiperfuncionantes de resolución quirúrgica. Si bien han sido diseñadas distintas estrategias de diagnóstico hay controversia alrededor de una serie de cuestiones. En el presente trabajo retrospectivo once (32%) de nuestros 34 pacientes presentaban masas adrenales hiperfuncionantes manifestadas por síndrome de Cushing subclínico en cuatro, feocromocitoma en tres, probable hiperaldosteronismo primario en dos y por hiperplasia adrenal congénita de origen tardío y carcinoma funcionante en los dos restantes. Las características de las imágenes por TAC y/o RM permitieron identificar los adenomas a la vez que decidir la cirugía tanto en dos pacientes con feocromocitomas bioquímicamente no funcionantes como en una paciente con un carcinoma adrenocortical primitivo, este diagnóstico también sugerido por un patrón mixto de hipersecreción de andrógenos y cortisol. En una paciente con síndrome de Cushing subclínico, hipertensa y diabética, ambas comorbilidades fueron resueltas por la cirugía. Los tumores no funcionantes fueron en su mayoría adenomas (87%), hallándose además histoplasmosis, pseudoquiste, hiperplasia suprarrenal idiopática y mielolipoma. Seis de los ocho pacientes operados tenían enfermedad maligna y/o hiperfuncionante. La condición asociada a los incidentalomas suprarrenales significó un amplio espectro de riesgo para los pacientes y reafirma la necesidad de una minuciosa evaluación clínica, bioquímica y de las imágenes a fin de adoptar conductas adecuadas.
Palabras clave: Tumores adrenales; Incidentaloma adrenal; Síndrome de Cushing subclínico
Abstract
A study of 34 cases of adrenal incidentaloma. Adrenal incidentaloma, a tumor discovered unexpectedly during imaging performed for non-adrenal related causes, has become a frequent clinical concern. Although in most cases they are benign and hormonally nonfunctioning, it is important to appropriately identify those few cases of malignant or hyperfunctioning lesions of surgical resolution. Although several proposals for an optimal diagnostic strategy have been designed, controversy over a series of questions still persists. In the present retrospective study we analyzed 34 patients with adrenal incidentaloma. Of these, 32% of the patients displayed hyperfunctioning pathologies that included subclinical Cushing's syndrome in four patients, probable primary hyperaldosteronism in two, late onset congenital adrenal hyperplasia in one, adrenocortical carcinoma in one and pheochromocytoma in three. CT and/or MRI permitted the identification of adenomas and were crucial to decide on surgery in two patients with nonfunctioning pheochromocytomas and in a patient carrying a primitive adrenocortical carcinoma, a diagnosis also suggested by a mixed pattern of hypersecretion of androgens and cortisol. In a diabetic and hypertensive patient with subclinical Cushing's syndrome both comorbidities were solved by surgery. Nonfunctioning tumors were mainly adenomas (87%) with individual cases of histoplasmosis, pseudocyst, idiopathic adrenal hyperplasia and mielolipoma. Six of the eight operated patients presented malignant and/or hyperfunctioning tumors. The pathology associated with incidentalomas represents a broad spectrum of risk for patients and reaffirms the necessity for a meticulous clinical, biochemical, and imaging evaluation in order to make appropriate decisions.
Key words: Adrenal gland tumors; Adrenal incidentaloma; Subclinical Cushing´s syndrome
El uso difundido de procedimientos de estudios por imágenes ha traído aparejado el descubrimiento casual de tumores suprarrenales en aproximadamente 2% de las tomografías computadas de abdomen de individuos sin evidencia clínica de enfermedad de dicha glándula1- 4. Estos tumores, a los que se conoce como incidentalomas adrenales, son por lo general lesiones benignas y no funcionantes; no obstante, en una minoría de pacientes pueden ser la expresión de enfermedad hiperfuncionante y/o maligna, potencialmente letal2, 3. Ocurre que, si bien los procedimientos bioquímicos permiten una eficaz discriminación entre las lesiones funcionantes y las no funcionantes, para el caso de los tumores no funcionantes, los estudios por imágenes brindan información solamente de orientación sobre su naturaleza benigna o maligna5, 6 y por más que el desarrollo de algoritmos posibilite habitualmente la toma de decisiones adecuadas, resulta inevitable la indicación de cirugía para algunos pacientes portadores de lesiones benignas. El panorama del incidentaloma adrenal (IA) se ha vuelto más intrincado aún en la medida en que recientes evidencias permitirían relacionar la sobreproducción hormonal subclínica por los incidentalomas adrenales con un incrementado riesgo cardiovascular. Vale decir, que por más que la problemática que plantea esta entidad viene siendo exhaustivamente analizada desde hace más de 20 años, subsiste polémica e incertidumbre acerca de la mejor conducta a adoptar en situaciones especiales.
El presente trabajo tiene por objetivo evaluar retrospectivamente nuestra experiencia con un grupo de pacientes con IA de modo de extraer conclusiones sobre la distribución de la patología causal y la utilidad de los procedimientos de diagnóstico y trapéuticos empleados.
Materiales y métodos
Se incluyeron 34 pacientes que nos consultaron entre 1993 y 2002, todos con nódulos de las glándulas suprarrenales de por lo menos 1 cm de diámetro descubiertos de manera casual por ecografía, tomografía computada (TAC) o resonancia magnética (RM). De acuerdo con la definición de incidentaloma3 adoptamos los criterios de exclusión siguientes: 1) presencia de manifestaciones clínicas ostensibles y específicas de disfunción suprarrenal al momento de la detección de la lesión, 2) antecedente o evidencia actual de neoplasia extra adrenal primitiva y 3) sintomatología localizada atribuible a un tumor.
Los motivos para la indicación inicial de los estudios por imágenes (dato obtenido en 30 pacientes) fueron los siguientes: dolor abdominal o toráxico característicos (n=12), cólico renal (n=4), neumopatía (n=3), colelitiasis (n=2), traumatismo (n=2), evaluación ginecológica (n=2), decaimiento general (n=1), hiperemesis (n=1), control de quiste renal (n=1), síndrome ascítico edematoso (n=1), hepatopatía (n=1).
La detección fue realizada por ecografía (n = 18), TAC (n = 15) o RM (n = 1) Toda vez que el IA fue descubierto por ecografía se obtuvo confirmación del hallazgo mediante TAC o RM.
En todos los pacientes fueron obtenidos datos relativos a las características de la imagen por TAC o RM incluyendo localización, diámetro mayor, forma, densidad, homogeneidad, márgenes y la presencia o no de invasión regional. Sobre la base de estas características, establecimos los siguientes diagnósticos presuntivos: adenoma, mielolipoma, quiste, carcinoma, hiperplasia bilateral o indefinido.
De acuerdo a criterios consensuados fueron caracterizadas como adenomas todas aquellas lesiones pequeñas, de suave contorno, márgenes netos, densidad baja y homogénea, que no hubieran mostrado crecimiento durante el seguimiento6, 7. Los quistes simples y los mielolipomas también tienen un diagnóstico específico por imágenes6.
Sobre la base de los antecedentes fue indagada la existencia de hipertensión arterial (HTA) y diabetes de tipo 2, y en 27 pacientes pudo calcularse el índice de masa corporal.
Los estudios bioquímicos realizados incluyeron: test de Nugent (cortisol sérico matinal luego de la administración de 1 mg de dexametasona la noche anterior) (n = 21), cortisol libre urinario (CLU) de 24 horas (n = 27), CLU de 22 a 23 horas (n = 21), sulfato de dehidroepiandrosterona (S-DHEA) (n = 24), adrenocorticotrofina (ACTH) (n = 13), 17-OH progesterona basal (n = 9), ácido vainillil mandélico (AVM) y/o catecolaminas urinarias (adrenalina y noradrenalina) (n = 31), kalemia (n = 26), actividad de renina plasmática (ARP) (n = 16, en 13 con medición simultánea de aldosterona sérica). Todos los pacientes hipertensos tuvieron registro de kalemia y/o del par aldosterona - ARP.
El criterio que adoptamos para el diagnóstico del síndrome de Cushing subclínico (SCs) incluyó la existencia de un test de Nugent no supresible (definido por un cortisol sérico ³ 3 µg/dl) conjuntamente con una incrementada cortisoluria (CLU-24 horas y/o de 22 a 23 horas)4, 8-10. Los demás resultados de las determinaciones bioquímicas realizadas fueron catalogados como normales, aumentados o disminuidos dependiendo del rango de referencia de cada laboratorio. El SDHEA representó una situación especial por cuanto la carencia de información sobre valores de referencia según sexo y edad ajustados para la metodología utilizada en cada paciente imposibilitó una clasificación confiable de los resultados como normales o disminuidos. El nivel de corte de la 17 OH-progesterona requerido para diagnóstico de hiperplasia adrenal congénita (HAC) fue de 5 ng/ml siguiendo criterios establecidos11.
En 27 pacientes se realizó un seguimiento con TAC durante un período de entre 2 y 96 meses. El diagnóstico definitivo se efectuó histológicamente en los pacientes operados y por las características de la imagen y la evolución en los demás.
Resultados
La población estudiada comprendió 34 pacientes, 17 varones y 17 mujeres, con una edad media de 53.4 años y una mediana de 59.5 años (rango: 8 a 74). El pico de frecuencia se ubicó en la 7a década, siendo el 71% de los pacientes mayores de 50 años (Fig. 1).

Fig. 1.- Distribución por edad de 34 pacientes con incidentaloma adrenal.
El 56% de los pacientes eran hipertensos, 20.6% diabéticos en tanto que 10 de 27 (37%) presentaban sobrepeso u obesidad.
La localizaci ón de la lesión adrenal fue: izquierda en 15 casos, derecha en 14 y bilateral en cuatro. El diáme- tro de las lesiones unilaterales fue de 3.5 ± 1.8 cm (rango: 1.5 a 8). Sobre la base de las características de la imagen establecimos que 24 pacientes presentaban adenomas unilaterales, 1 un mielolipoma, 1 un quiste, 1 un carcinoma (confirmado histológicamente), tres hiperplasias bilaterales (una de ellas nodular). De los cuatro pacientes con diagnóstico indefinido por TAC tres tenían un feocromocitoma y uno histoplasmosis. El diámetro de los adenomas, fue de 3.1 ± 0.8 cm (rango 1.5 a 4.5) mientras que el de los no-adenomas unilaterales (n = 6) fue de 6.2 ± 1.5 cm (rango 5 a 8) (p <0.0001). Los adenomas constituyeron el 80% de los incidentalomas unilaterales y la totalidad del subgrupo de masas unilaterales menores de 4 cm. Los cinco adenomas con diámetro ³ 4 cm representaron el 21% de los adenomas y el 45% de las masas unilaterales de por lo menos 4 cm.
Establecimos que en 23/34 pacientes (68%) las lesiones eran no funcionantes en tanto que en once (32%) eran hiperfuncionantes (Tabla 1). En una paciente portadora de un adenoma no se explor ó el eje hipotálamohipófiso- adrenal; no obstante, a causa de haberse diagnosticado HAC de comienzo tardío, por un nivel (confirmado) de 17-OH-progesterona de 12.3 ng/ml, su nódulo suprarrenal fue clasificado como hiperfuncionante.
TABLA 1.- Distribución de 34 pacientes con incidentaloma adrenal según su condición funcional y etiología

Los datos m ás relevantes de nuestros tres pacientes con feocromocitoma están detallados en la Tabla 2. Destacamos que en dos de ellos los resultados de los estudios bioquímicos, dentro del rango de referencia, no posibilitaron el diagnóstico.
TABLA 2.- Feocromocitomas presentados como incidentaloma

El resultado del test de Nugent fue de 5.9 ± 8.0 µg /dl (1.0 a 31.4); el del CLU de 24 horas de 68.1 ± 32.8 µg/ 24 horas (rango: 0 a 153) y el del CLU de 22 a 23 horas de 31.8 ± 47.8 ng/mg de creatinina (rango: 4.7 a 225). En 10 de 12 pacientes en los que no fue realizado el test de Nugent el diagnóstico de SCs quedó descartado por un CLU dentro del rango normal (ver pacientes y métodos). Unicamente fueron hallados valores incrementados de CLU en los cuatro pacientes con SCs (cuyas características se detallan en la Tabla 3). Una paciente con SCs, diabética e hipertensa, desarrolló insuficiencia suprarrenal transitoria luego de adrenalectomía unilateral por adenoma; junto con la posterior normalización de la dinámica de su eje hipotálamo-hipófiso-adrenal se produjo también normalización de las glucemias y niveles tensionales.
TABLA 3.- Evaluación bioquímica de los pacientes con incidentaloma asociado a síndrome de Cushing subclínico
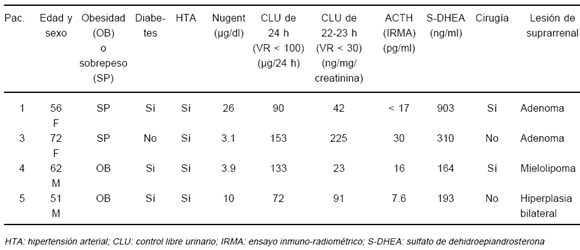
En 12 de los 20 pacientes portadores de ANF fue realizado un test de Nugent; seis de ellos presentaron valores entre 1 y 2.9 µg /dl y en otros seis fueron ³ 3 µg /dl, aunque todos ellos tenían cortisoluria normal.
El nivel promedio de S-DHEA fue de 702 ± 956 ng/ml (rango: 70 a 5000); solamente fueron observados valores incrementados en la paciente portadora de un carcinoma suprarrenal, en quien un nivel de S-DHEA de 5000 ng/ml se consideró incuestionablemente elevado a sus 69 años. Esta paciente tenía además concentraciones francamente incrementadas de testosterona (4.6 ng/ml) y de androstenodiona (5 ng/ml) y un test de Nugent definidamente patológico (cortisol post dexametasona: 31.4 µg/dl).
De los 19 pacientes hipertensos dos ten ían un feocromocitoma, dos hiperaldosteronismo primario, en 4 se diagnosticó SCs, en tanto que 11 (58%) eran portadores de adenomas no funcionantes (ANF). Sobre la base de una elevada relación aldosterona/ARP, en dos pacientes se realizó el diagnóstico de hiperaldosteronismo primario (HAP), en un caso asociado a un adenoma y en el otro a hiperplasia bilateral (Tabla 4). Si bien no se pudo realizar prueba de supresión para su confirmación, el diagnóstico estuvo convalidado por la presencia de franca hipokalemia en un caso y por una respuesta clínica muy favorable a bajas dosis de espironolactona en ambos pacientes.
TABLA 4.- Características de los pacientes con hiperaldosterenismo primario

De los 23 pacientes con incidentalomas no funcionantes 20 (87%) presentaron un adenoma (en uno de ellos asociado a mielolipoma descubierto durante la biopsia quir úrgica); los tres pacientes restantes tenían respectivamente histoplasmosis adrenal bilateral, pseudoquiste e hiperplasia adrenal bilateral idiopática.
De los cuatro pacientes con lesiones bilaterales uno present ó SCs y el otro hiperaldosteronismo primario, en ambos casos asociados a hiperplasia bilateral. Los dos restantes presentaron lesiones no funcionantes (hiperplasia bilateral idiopática e histoplasmosis).
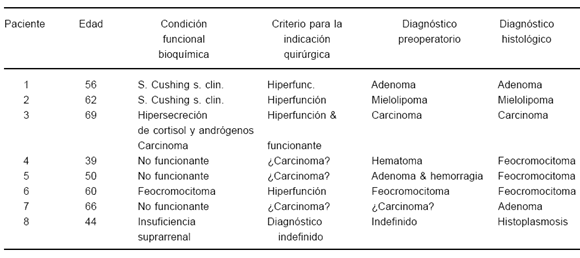
Ocho pacientes fueron operados (Tabla 5). En cuatro el diagn óstico bioquímico de hiperfunción suprarrenal fue decisivo, o cuanto menos relevante, para la indicación de cirugía: SCs en dos pacientes, feocromocitoma en otro e hipersecreción mixta de cortisol y andrógenos en la paciente con carcinoma. En tres de cuatro pacientes con masas bioquímicamente no funcionantes, la cirugía fue indicada sobre la base de su tamaño (³ 5 cm) y de otras características de la imagen, atípicas para un adenoma. En la cuarta paciente, la cirugía obedeció al crecimiento del nódulo de 4.4 cm con la apariencia de un adenoma, diagnóstico que fue confirmado por la histología. En los cinco pacientes en los que la vía de abordaje fue laparoscópica no se presentaron complicaciones. En una paciente con feocromocitoma el procedimiento debió convertirse a laparotomía.
TABLA 5.- Pacientes con incidentalomas operados
En 26 pacientes inicialmente no operados, se realiz ó un seguimiento mediante TAC durante un período de 2 a 96 meses. En 23 (89%) el tamaño se mantuvo estable, observándose crecimiento en una paciente y disminución de tamaño en dos, todos portadores de un ANF.
Discusión
El incidentaloma suprarrenal es definido como un tumor de dicha glándula no sospechado con anterioridad al procedimiento por imágenes que condujo a su descubrimiento. No constituye una entidad patológica única y, por el contrario, incluye múltiples posibles etiologías las que representan un espectro de riesgo potencial para los pacientes.
Tal como está comunicado, observamos en nuestra serie de pacientes un franco predominio en individuos de ambos sexos mayores de 50 años, grupo de edad en el que la prevalencia de IA es de hasta 7%1, 4. Estos datos concuerdan con los hallazgos de autopsia, según los cuales los adenomas suprarrenales son hallados con una frecuencia creciente -de hasta 8.9%- a medida que progresa la edad2.
El 32% de pacientes con masas hiperfuncionantes de nuestra serie supera ampliamente el 15% comunicado recientemente en una extensa serie multicéntrica4; esto podría guardar relación con el hecho de que hayamos decidido incluir en esta categoría a dos pacientes que presentaban, además de criterios bioquímicos de SCs, signos clínicos sutiles e inespecíficos de hipercortisolismo. La distribución de la patología causal y de la condición funcional de los incidentalomas está íntimamente vinculada a su definición, a los criterios de inclusión y de exclusión adoptados y a la metodología de estudio empleada2. Si bien por definición no debieran existir signos o síntomas del síndrome de Cushing en pacientes con incidentaloma, en coincidencia con Reincke9 creemos que es admisible que un cuestionario más detallado o un cuidadoso segundo examen físico puedan revelar evidencias sutiles de exceso hormonal las que, para el juicio de un observador experimentado, podrían no tener la jerarquía suficiente como para decidir apartar a estos tumores del grupo de incidentalomas. Además, dichas manifestaciones suelen incluir a los componentes del síndrome metabólico, los que son significativamente más frecuentes en los pacientes con incidentaloma que en la población general y por lo tanto de baja especificidad.
Nuestros tres casos de feocromocitomas representaron un 8.6% de la serie, en coincidencia con alrededor de un 10% comunicada por otros autores2, 4, 12. El hallazgo de niveles aumentados de catecolaminas urinarias y de AVM en sólo uno de nuestros pacientes con feocromocitoma podría atribuirse al hecho de no haber empleado las determinaciones bioquímicas más sensibles, como son las metanefrinas urinarias fraccionadas12 o las plasmáticas13, 14. En efecto, por más que fue comprobada la existencia de un feocromocitoma no funcionante a causa de la síntesis alterada de catecolaminas15, puede ocurrir que la condición de "no funcionante" sea tan solo aparente y debida, ya sea a una secreción tumoral episódica o a la insuficiente sensibilidad de los procedimientos bioquímicos utilizados para su detección13. El riesgo considerable que significa que un feocromocitoma pase irreconocido tiene que ver con una incrementada mortalidad derivada tanto de complicaciones perioperatorias (prevenibles mediante tratamiento médico) como de eventos cardiovasculares o cerebrovasculares, frecuentes en la historia natural del feocromocitoma no tratado4,16. Afortunadamente, así ocurra en individuos normotensos y asintomáticos, por lo general un feocromocitoma es puesto de manifiesto por la investigación bioquímica2, 4. Recomendamos que en todo paciente portador de un IA sea investigada bioquímicamente la presencia de un feocromocitoma y que, aun cuando no se demuestre hipersecreción hormonal, ante evidencia de otro orden que sugiera este diagnóstico, se proceda a su extirpación quirúrgica sin postergaciones2. En coincidencia con otros autores encontramos además que, tuvieran o no expresión bioquímica, todos los feocromocitomas tenían por TAC un diámetro de por lo menos 5 cm con una densidad intermedia4 y por RM en T-2 la hiperintensidad característica17. El gran tamaño unido a la heterogeneidad de la imagen hace que los feocromocitomas puedan ser confundidos con carcinomas2, 5, justificándose estudios altamente específicos como la centelleografía con meta-yodo-bencilguanidina (MIBG) para hacer el diagnóstico diferencial prequirúrgico18.
El SCs ha concitado renovado interés desde que se lo asocia con un perfil de incrementado riesgo cardiovascular2, 19 - 21. Su prevalencia varía dentro de un amplio rango y ello obedecería a que no se dispone de un verdadero "gold standard" de diagnóstico22. Así, por ejemplo, en circunstancias en las que el diagnóstico dependió de la presencia de cortisol no supresible por dexametasona asociado a hipercortisoluria, su frecuencia estuvo en el rango de 5 a 12%8, 23; en cambio, cuando se emplearon procedimientos adicionales llegó hasta 24%10. En dos de nuestros pacientes con SCs la hipercortisoluria se puso de manifiesto en la muestra de 22 a 23 horas pero no en la de 24 horas. Esto está de acuerdo con datos de otros autores refiriendo valores ligeramente elevados de CLU en la muestra vespertina, a pesar de resultados normales en la determinación de 24 horas en la mayoría de sus pacientes con incidentaloma24. Coincidimos en recomendar que todo paciente portador de un IA sea evaluado inicialmente con un test de Nugent3, 25; ante un resultado anormal corresponderá que sea medido además el CLU de 24 horas y, de ser factible, en una muestra vespertina de 1 hora. En nuestra experiencia la hipercortisoluria se circunscribió a los pacientes con SCs; para otros autores, en cambio, la hipercortisoluria aislada constituyó un hallazgo relativamente frecuente, de cuestionable valor como único indicador de hipersecreción glucocorticoidea4, 9. Los niveles normales de ACTH (IRMA) que hallamos en dos de nuestros pacientes con SCs -una observación también de otros autores26- estuvieron así mismo presentes en algunos de nuestros pacientes con síndrome de Cushing clínicamente manifiesto de origen primitivamente suprarrenal (datos no publicados por los autores). Debe recordarse que, a diferencia de la TSH, la mayoría de los ensayos para ACTH no tienen una sensibilidad suficiente para discriminar confiablemente entre los valores verdaderamente descendidos y los normales27. La no supresibilidad por dexametasona manifestada en seis de nuestros pacientes con ANF sugiere, tal como ha sido comunicado, algún grado de autonomía de estos tumores en la secreción de cortisol23. Si bien no se descarta que se trate de resultados falsos positivos, es factible que la secreción de cortisol haya sido verdaderamente no supresible así no se expresara a través de un nivel incrementado de CLU. Esta disociación se explica admitiendo que la hipersecreción de cortisol por los adenomas adrenales conformaría un todo continuo sin línea de corte neta entre la anormalidad leve y la completamente patológica, y que en el proceso de desarrollo de la autonomía secretoria de cortisol la hipercortisoluria constituiría un fenómeno de aparición tardía9, 22, 23, 26. El criterio utilizado para el diagnóstico de SCs sería entonces arbitrario y sólo permitiría reconocer los grados mayores de hipersecreción subclínica de cortisol10, 26. Por más que los expertos continúen poniendo énfasis en la necesidad de refinar los estudios bioquímicos para establecer con mayor precisión el diagnóstico del SCs, debe recordarse que esta entidad no ha sido aún adecuadamente caracterizada, siendo desconocida hasta ahora su historia natural. Si bien el tratamiento quirúrgico ha resultado efectivo para resolver comorbilidades en casos aislados3, tal como sucedió con una de nuestras pacientes, se desconoce en qué medida el SCs tiene consecuencias a largo plazo y si el tratamiento destinado a revertir el exceso sutil de glucocorticoides ofrece verdaderamente beneficios25. Hasta tanto no sea factible reconocer un punto a partir del cual la autonomía secretoria de cortisol sea capaz de producir morbilidad clínica significativa, no vamos a estar en condiciones de establecer cuán agresivo debería ser nuestro enfoque diagnóstico y terapéutico27.
Distintos autores refieren la existencia de una alta proporción de pacientes con IA con niveles descendidos de S-DHEA2, 4. Se ha postulado que los mismos podrían constituir un indicador de la autonomía secretoria de cortisol; sin embargo, subsiste controversia alrededor de su significado y de los mecanismos por los que tienen lugar 28, 29. Aunque no pudimos diferenciar los valores normales de los descendidos de S-DHEA (ver sección Materiales y métodos), un nivel definidamente incrementado de S-DHEA asociado a la hipersecreción subclínica de cortisol contribuyó a establecer el diagnóstico de carcinoma adrenal en una paciente. Este patrón mixto de hipersecreción de cortisol y andrógenos es observado frecuentemente en casos de carcinomas hiperfuncionantes, a la vez que son sumamente infrecuentes los adenomas productores de andrógenos en el adulto2. Si bien, al igual que otros, el S-DHEA no constituye un marcador bioquímico totalmente confiable de carcinoma adrenal, por tratarse de una determinación accesible y económica y por encontrarse sus niveles elevados en aproximadamente 25% de estos pacientes se justifica que continúe siendo medido2-4. El pronóstico de los carcinomas suprarrenales es pobre, estando la sobrevida claramente vinculada a la extensión de la enfermedad y a la posibilidad de una resección completa30; es por este motivo, que optamos por indicar la cirugía toda vez que se susciten dudas sobre la condición de benignidad de un IA.
La hiperplasia adrenal congénita (HAC), diagnosticada en una de nuestras pacientes, se asocia frecuentemente con tumores suprarrenales uni o bilaterales31; ello se explicaría por un exceso de estimulación por ACTH, tal como ocurre en el síndrome de Cushing ACTH-dependiente. Los pacientes con IA asociada a HAC presentaron casi siempre enfermedad benigna31, 32. El diagnóstico de HAC en el contexto del IA ofrece dificultades, justificándose su investigación exhaustiva únicamente sobre la base de criterios clínicos o ante la existencia de masas adrenales bilaterales27, 33, 34.
La proporción de HAP entre los paciente con IA es de 1 al 4%2-4. Se ha atribuido esta baja frecuencia a que habitualmente se excluye de la condición de incidentaloma a los pacientes gravemente hipertensos con hipokalemia35. Aun así llama la atención esta baja prevalencia si se tiene en cuenta que en series no seleccionadas el síndrome de Conn normokalémico es diagnosticado en 6 a 10% de los hipertensos36. En nuestra serie retrospectiva es factible que hayamos incurrido en subdiagnóstico en la medida en que no fue medido sistemáticamente el par aldosterona-ARP.
Es destacable la alta proporción de hipertensos entre nuestros pacientes (56%), de los cuales 58% presentaba un ANF. Según grandes series, la prevalencia de hipertensión arterial entre los paciente con IA es de 40%, superior al 20 a 30% correspondiente a la población general. En la mayoría de los casos está asociado al síndrome metabólico, condición que también explicaría la frecuencia mayor de obesidad y diabetes2- 4, 20.
Entre los pacientes con enfermedad no funcionante hubo franco predominio de adenomas, los que representaron el 87% de este subgrupo y el 59% de la serie total. Es en los pacientes con masas no funcionantes en los que se ponen de manifiesto las limitaciones de los procedimientos de diagnóstico, y es el tamaño el dato que se toma en cuenta como primera aproximación para diferenciar las lesiones benignas de las malignas. El diámetro significativamente mayor de los no adenomas, comparado con el de los adenomas existentes en nuestra serie de pacientes era de esperar y coincide con informes publicados2. Una proporción del 45% de adenomas entre las masas unilaterales de por lo menos 4 cm tampoco es de extrañar si se tiene en cuenta la no infrecuente presentación de grandes adenomas como incidentalomas adrenales2. En cambio, los carcinomas menores de 4 cm son muy raros, e infrecuentes los menores de 6 cm. La frecuencia relativa de los grandes adenomas se ha calculado en alrededor de 60 veces la de los carcinomas3 y en una serie numerosa, entre las masas adrenales mayores de 6 cm, los carcinomas representaron el 25%2; vale decir que por más que el riesgo de malignidad se acreciente con el tamaño, continúa siendo mayor la probabilidad de que los grandes incidentalomas sean benignos y no malignos3. Las líneas de corte del diámetro propuestas para decidir entre la cirugía y una conducta expectante varían entre 2.5 y 6 cm4, sin embargo, ninguna tiene una sensibilidad y especificidad de 100% para diferenciar las lesiones benignas de las malignas. Nosotros, en coincidencia con otros autores, hemos adoptado el diámetro de 5 cm37, haciendo hincapié en que, cuando se trate de lesiones menores de 5 cm, sean consideradas otras características de la imagen para la toma de decisiones3 (Fig. 2).

Fig. 2.- Algoritmo para el enfoque del paciente con incidentaloma adrenal.
En ninguno de nuestros pacientes indicamos la realización de patología del aspirado con aguja fina (PAAF). Este es un procedimiento que tiene infrecuente aplicación en pacientes portadores de un IA, por no estar exento de complicaciones y porque un diagnóstico citológico de benignidad no excluye un carcinoma suprarrenal2, 25. La reconocida eficacia de la citología para diferenciar adenomas de metástasis38 no justifica su empleo en el contexto del IA dado que en estos pacientes, sin antecedentes oncológicos, es muy remota la posibilidad de una metástasis39. Los nuevos desarrollos de técnicas por imágenes (TAC y RM) podrían tener como resultado una restricción aún mayor de las indicaciones de la PAAF40 - 42.
Nuestros tres pacientes con masas adrenales bilaterales representaron el 8.6% del total, una proporci ón comprendida dentro del rango de 2 a 10% referida en la literatura3,4, 43. En uno de ellos, el desarrollo de hiperplasia adrenal nodular bilateral asociada a SCs podría relacionarse hipotéticamente con la expresión aberrante de receptores de membrana y la activación patológica de una serie de proteínas G acopladas a los mismos, tal como fuera descrito por Lacroix44.
Los incidentalomas bilaterales no funcionantes pueden producir insuficiencia suprarrenal, condici ón que debiera ser diagnosticada lo antes posible. Este fue el caso de nuestro paciente con histoplasmosis, quien en el transcurso de su evolución desarrolló insuficiencia suprarrenal la que, tal como está referido, revirtió por completo luego del tratamiento médico específico45.
Los criterios adoptados para la decisi ón de la cirugía fueron redituables en términos de riesgo/beneficio ya que casi todos los pacientes con masas hiperfuncionantes se beneficiaron por ella, lo mismo que tres de cuatro pacientes con nódulos bioquímicamente no funcionantes (dos con feocromocitoma y otro producido por histoplasmosis). La cirugía laparoscópica, en las manos de cirujanos experimentados, constituye un procedimiento más aceptable que la laparotomía dada su tasa más baja de complicaciones, menor tiempo operatorio y de internación, menor dolor postoperatorio y rápida recuperación de la función vesical25. No obstante, si bien esta técnica ofrece reales ventajas y va adquiriendo progresiva difusión, subsiste controversia alrededor de algunas de sus indicaciones particularmente cuando se trata de grandes tumores, más probablemente malignos, en los que la resección es más dificultosa e involucra el riesgo de rotura o de exéresis incompleta46. Consecuentemente, algunos recomiendan la cirugía abierta para los tumores adrenales de 6 cm o mayores con significativa probabilidad de cáncer46, 47. Para otros, el diámetro no constituiría un factor limitante siempre que el tumor esté confinado a los límites de la adrenal, existiendo siempre la posibilidad de la conversión a laparotomía toda vez que se constate invasión o ante la presencia de complicaciones. En la medida en que por el momento no se cuenta con resultados de estudios prospectivos comparativos, ambos procedimientos se consideran aceptables, lo mismo que una u otra vía de abordaje laparoscópico (transabdominal o retroperitoneal)25.
Como ocurre habitualmente, la gran mayor ía de nuestros pacientes que tuvieron control evolutivo, mantuvieron estable el tamaño de sus lesiones2. Datos derivados de series relativamente pequeñas indican que 3 a 20% de los incidentalomas no operados incrementan su tamaño, que en 3 a 4% éste se reduce y que menos del 20% desarrollan anormalidades bioquímicas (más frecuentemente hipersecreción de cortisol) cuando son seguidos durante hasta 10 años2, 48. El SCs ha sido incriminado como un factor de riesgo para el desarrollo de síndrome de Cushing manifiesto49, sin embargo, esta evolución parece ser infrecuente48. La escasez de estudios analizando de modo prospectivo la evolución de pacientes con masas adrenales no tratadas impide que puedan formularse recomendaciones específicas para el seguimiento, no obstante, los datos disponibles hacen pensar que el riesgo de un carcinoma es bajo25 y que es muy probable que aquellos pacientes cuyo diámetro tumoral permanezca estable al año de observación y que no manifiesten hipersecreción hormonal en el lapso de 4 años, podrían no requerir estudios ulteriores2,25, 48.
En conclusión:
- Los IA de nuestra serie representaron un amplio espectro de riesgo para los pacientes.
- Los tests bioquímicos fueron útiles para reconocer incidentalomas hiperfuncionantes, si bien su sensibilidad resultó insuficiente para diagnosticar feocromocitoma en dos pacientes.
- En una paciente con SCs el tratamiento quirúrgico repercutió favorablemente sobre sus comorbilidades.
- Los pacientes con ANF podrían presentar alteraciones sutiles de la dinámica secretoria de cortisol.
- Un nivel incrementado de SDHEA contribuyó a reconocer un carcinoma suprarrenal.
- El diámetro, lo mismo que otras características de la imagen por TAC fueron útiles para el diagnóstico de adenoma, posteriormente corroborado por la evolución o la histología
- La imagen por TAC permitió diagnosticar un quiste y un mielolipoma y sugirió fuertemente el diagnóstico de carcinoma adrenocortical.
- La RM contribuyó a decidir la cirugía en el caso de una paciente normotensa y asintomática, portadora de un feocromocitoma de 5 cm con determinaciones bioquímicas normales y diagnóstico radiológico incierto por TAC.
- Los criterios adoptados, plasmados en nuestro algoritmo, permitieron evitar la cirugía en la gran mayoría de los pacientes portadores de lesiones benignas e indicarla en todos aquellos con lesiones malignas o con tumores hiperfuncionantes.
- Durante el seguimiento por imágenes no se modificó el diagnóstico original en ninguno de nuestros pacientes; el crecimiento tumoral no estuvo asociado a malignidad.
Bibliografía
1. Herrera MF, Grant CS, van Heerden JA, Sheedy PF, Ilstrup DM. Incidentally discovered adrenal tumors: an institutional perspective. Surgery 1991; 110: 1014-21.
2. Mansmann G, Lau J, Balk E, Tothberg M, Miyachi Y, Bornstein SR. The clinically inapparent adrenal mass: update in diagnosis and management. Endocrine Rev 2004; 25: 309-40.
3. Copeland PM. The incidentally discovered adrenal mass: an update. Endocrinologist 1999; 9: 415-23.
4. Mantero F, Terzolo M, Arnaldi G, et al. A survey on adrenal incidentaloma in Italy. J Clin Endocrinol Metab 2000; 85: 637-44.
5. Reinig JW, Doppman JL, Dwyer AJ, Frank J. MRI of indeterminate adrenal masses. AJR Amer J Roentgenol 1986; 147: 493-6.
6. Gross MD, Falke THM, Shapiro B, Sandler MP. Adrenal gland. In: Sandler MP, Patton JA, Gross MD, Shapiro B, Falke THM (eds). Endocrine Imaging. Norwalk, Connecticut/ San Mateo/California: Appleton & Lange, 1992, ch. 11, p 271-349.
7. Korobkin M, Francis IR, Kloos RT, Dunnick NR. The incidental adrenal mass. Radiol Clin North Am 1996; 34: 1037-55.
8. Ross NS. Epidemiology of Cushing´s syndrome and subclinical disease. Endocrinol Metab Clin North Am 1994; 23: 539-46.
9. Reincke Martin. Subclinical Cushing´s syndrome. Endocrinol Metab Clin North Am 2000; 29: 43-56.
10. Rossi R, Tauchmanova L, Luciano A, et al. Subclinical Cushing´s syndrome in patients with adrenal incidentaloma: clinical and biochemical features. J Clin Endocrinol Metab 2000; 85: 1440-8.
11 Azziz R, Dewailly D, Owerbach D. Nonclassic adrenal hyperplasia: current concepts. J Clin Endocrinol Metab 1994; 78: 810-815.
12. Kudva YC, Young WF, Thompson GB, Grant CS, van Heerden JA. Adrenal incidentaloma: an important component of the clinical presentation spectrum of benign sporadic adrenal pheochromocytoma. Endocrinologist 1999; 9: 77-80.
13. Raber W, Raffesberg W, Kmen, E., et al. Pheochromocytoma with normal urinary and plasma catecholamines but elevated plasma free metanephrines in a patient with adrenal incidentaloma. Endocrinologist 2000; 10: 65-8.
14. Lenders JW, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocitoma: which test is best? JAMA 2002; 287: 1427-34.
15. Mannelli M, Pupilli C, Lanzillotti R, et al. A nonsecreting pheochromocytoma presenting as an incidental mass. Report on a case. J Endocrinol Invest 1993; 16: 817-22.
16. St. John Sutton, MG, Sheeps SG, Lie JT. Prevalence of clinically unsuspected pheocromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc 1981; 56: 354-60.
17. Fink IJ, Reinig JW, Dwyer AJ, Doppman JL, Linehan WM, Keiser HR. MR imaging of pheochromocytomas. 1985; J Comp Assist Tomogr 9: 454-8.
18. Shapiro B, Copp JE, Sisson JC, Eyre PL, Wallis J, Beierwaltes WH. Iodine-131 metaiodobenzilguanidine for the locating of suspected pheochromocytoma: experience of 400 cases. J Nucl Med 1985; 25: 576-85.
19. Terzolo M, Pia A, Ali A, et al. Adrenal incidentaloma: a new cause of the metabolic syndrome? J Clin Endocrinol Metab 2002; 87: 998-1003.
20. Fernandez-Real JM, Gonzalbez J, Ricart W. Metabolic abnormalities in patients with adrenal incidentaloma. J Clin Endocrinol Metab 2000; 85: 1440-8.
21. Tauchmanova L, Rossi R, Biondi B, et al. Patients with subclinical Cushing´s syndrome due to adrenal adenoma have increased cardiovascular risk. J Clin Endocrinol Metab 2002; 87: 4872-8.
22. Aron D. Adrenal incidentalomas and glucocorticoid autonomy (Commentary). Clin Endocrinol 1998; 49: 157-8.
23. Tsagarakis S, Roboti C, Kokkoris P, Vasiliou V, Alevizaki C, Thalassinos N. Elevated post-dexametasone suppression cortisol concentrations correlate with hormonal alterations of the hypothalamo-pituitary adrenal axis in patients with incidentalomas. Clin Endocrinol 1998; 49: 165-71.
24. Laudat MH, Billaud L, Thomopoulos P, Vera O, Yllia A, Luton JP. Evening urinary free corticoids: a screening test in Cushing´s syndrome and incidentally discovered adrenal tumors. Acta Endocrinol 1988; 119: 459-64.
25. Grumbach MM, Biller BM, Braunstein GD, Campbell KK, Carney JA, Godley PA, et al. Management of the clinically inapparent adrenal mass ("incidentaloma"). Ann Intern Med 2003; 138: 424-29.
26. Tanabe A, Naruse M, Nishikawa T, et al. Autonomy of cortisol secretion in clinically silent adrenal incidentaloma. Horm Metab Res 2001; 33:444-50.
27. Young, WF. Jr. Management Approaches to Adrenal Incidentalomas. A view from Rochester, Minnesota. Endocrinol Metab Clin North Am 2000;
29: 159-84.
28. Terzolo M, Osella G, Ali A, et al. Subclinical Cushing´s syndrome in adrenal incidentaloma. Clin Endocrinol 1998; 48: 89-97.
29. Bencsik Z, Szabolcs I, Kovacs Z, et al. Low dehydroepiandrosterone sulfate (DHEA-S) is not a good predictor of hormonal activity in non selected patients with incidentally detected adrenal tumors. J Clin Endocrinol Metab 1996; 81: 1726-9.
30. Stratakis CA, Chrousos GP. Adrenal cancer. Endocrinol Metab Clin North Am 2000; 29: 15-25.
31. Jaresch S, Kornely E, Kley HK, Schlaghecke R. Adrenal incidentaloma and patients with homozygous or heterozygous congenital adrenal hyperplasia. J Clin Endocrinol Metab 1992; 74: 685-9.
32. Merke DP, Bornstein S, Braddock D, Chrousos GP. Adrenal limphocytic infiltration and adrenocortical tumors in patients with 21-hydroxylase deficiency. N Eng J Med 1999; 340: 1121-2.
33. Del Monte P, Bernasconi D, Bertolazzi L, et al. Increased 17a-hydroxyprogesterone response to ACTH in silent adrenal adenoma: cause or effect? Clin Endocrinol 1995; 42: 273-7.
34. Kjellman M, Holst M, Backdahl M, Larsson C, Farnebo LO, Wedell A. No overrepresentation of congenital adrenal hyperplasia in patients with adrenocortical tumors. Clin Endocrinol 1999; 50: 343-6.
35. Mantero F and Arnaldi G. Management Approaches to Adrenal Incidentalomas. A view from Ancona, Italy. Endocrinol Metab Clin North Am 2000; 29: 107-24.
36. Gordon RD, Stowasser M, Tunny TJ, Klemm SA, Rutherford JC. High incidence of primary aldosteronism in 199 patients referred with hypertension. Clin Exp Pharmacol Physiol 1994; 4: 315-8.
37. Bornstein SR, Stratakis CA, Chrousos GP. Adrenocortical tumors: recent advances in basic concepts and clinical management. Ann Intern Med 1999; 130: 759-71.
38. Katz RL, Shirkhoda A. Diagnostic approach to incidental adrenal nodules in the cancer patient. Results of the clinical, radiological, and fine needle aspiration study. Cancer 1985; 55: 1995-2000.
39. Lee JE, Evans DB, Hickey RC, et al. Unknown primary cancer presenting as an adrenal mass: frequency and implications for diagnostic evaluation of adrenal incidentalomas. Surgery 1998; 124: 1115-22.
40. Caoili EM, Korobkin M, Francis IR, et al. Adrenal masses: characterization with combined unenhanced and dilayed enhanced CT. Radiology 2002; 222: 629-33.
41. Bilbey JH, McLoughlin RF, Kurkjian PS, Wilkins GE, Chan NH, Schmidt N. et al. MR imaging of adrenal masses: value of chemical-shift imaging for distinguishing adenomas from other tumors. AJR Amer J Roentgenol 1995; 164: 637-42.
42. Heinz-Peer G, Honigschnabl S, Niederle B, Lechner G. Characterization of adrenal gland space-occupying lesions. Can diagnostic imaging replace biopsy? Radiologe1999; 7: 578-83.
43. Latronico AC, Chrousos GP. Extensive personal experience: adrenocortical tumors. J Clin Endocrinol Metab 1997; 5: 1317-24.
44. Bourdeau I, D´Amour P, Hamet P, Boutin JM, Lacroix A. Aberrant membrane hormone receptors in incidentally discovered macronodular adrenal hyperplasia with subclinical Cushing´s syndrome. J Clin Endocrinol Metab 2001; 86: 5534-40.
45. Washburn RG, Bennett JE. Reversal of adrenal glucocorticoid dysfunction in a patient with a disseminated histoplasmosis. Ann Intern Med 1989; 110: 86-7.
46. Winfield HN, Hamilton BD, Bravo EL, Novick AC, Laparoscopic adrenalectomy: the preferred choice? A comparison to open adrenalectomy. J Urol 1998; 160: 325-9.
47. Wells SA, Merke DP, Cutler GB Jr, Norton JA, Lacroix A. The role of laparoscopic surgery in adrenal disease. J Clin Endocrinol Metab 1998; 83: 3041-9.
48. Barzon L, Scaroni C, Sonino N, Fallo F, Paoletta A, Boscaro M. Risk factors and long-yerm follow-up of adrenal incidentalomas 1999; J Clin Endocrinol Metab 84: 520-6.
49. Barzon L, Fallo F, Sonino N, Boscaro M. Development of overt Cushing's syndrome in patients with adrenal incidentaloma. Eur J Endocrinol 2002 ; 146: 61-6. [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ]
Recibido: 31-08-2006
Aceptado: 19-03-2007

