










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.67 n.5 Buenos Aires sep./oct. 2007
Feocromocitoma asociado a neurofibromatosis de von Recklinghausen
Ramón N. Herrera, Julio A. Miotti, Claudio M. Fuentes, Silvia P. Robles, Jesús M. Amenabar, Héctor L. Luciardi
Departamento de Clínica Médica, Hospital Centro de Salud Zenón J. Santillán, Tucumán
Dirección postal: Dr. Ramón Nicasio Herrera, Marcos Paz 796 3D, 4000 Tucumán, Argentina Fax: (0381) 430-6518 e-mail: nicasioherrera@arnet.com.ar
Resumen
El feocromocitoma es un tumor glandular adrenal secretor de hormonas epinefrina y norepinefrina, responsables de regular la frecuencia cardíaca y la presión arterial, entre otras funciones. Este tumor puede ocurrir solo o en combinación con otros desórdenes; los factores genéticos y ambientales juegan un rol clave en su aparición. La neurofibromatosis tipo 1 (NF-1) es un desorden genético frecuente que se hereda en forma autosómica dominante, caracterizado por la formación de neurofibromas (tumores que involucran los nervios tisulares) en piel, tejido subcutáneo, nervios craneales y espinales. La NF-1 se diagnostica generalmente con el examen físico. No existe un tratamiento curativo para la NF-1, pero hay modos de tratar algunas de sus complicaciones. La hipertensión arterial en la neurofibromatosis causada por un feocromocitoma es extremadamente rara con una incidencia de menos del 1% en menores de 10 años y en adultos jóvenes. Presentamos el caso clínico de una mujer joven con hipertensión de reciente diagnóstico, con la infrecuente asociación de neurofibromatosis y feocromocitoma. Discutimos los mecanismos fisiopatológicos subyacentes y sus implicancias clínicas.
Palabras clave: Feocromocitoma; Neurofibromatosis; Tratamiento; Hipertensión arterial
Abstract
Pheochromocytoma associated with von Recklinghausen neurofibromatosis. A pheochromo cytoma is an adrenal gland tumor that secretes epinephrine and norepinephrine hormones, and is responsible for regulating heart rate and blood pressure, among other functions. The condition can occur alone or in combination with other disorders, and genetic and environmental factors play a key role. Neurofibromatosis- 1 (NF-1) an inherited "autosomal dominant" disorder is one of the most common genetic disorders, characterized by formation of neurofibromas (tumors involving nerve tissue) in the skin, subcutaneous tissue, cranial and spinal root nerves. NF1 generally is diagnosed by physical examination. There is no cure for NF1, but there are ways to treat some of its effects. Neurofibromatosis arterial hypertension caused by pheochromocytoma is extremely rare, less frecuent than 1% in childrens less than 10 years old, and young adults. We present a case of an extremely infrequent association between neurofibromatosis and a pheochromocytoma in a young woman with a newly diagnosed hypertension. We discuss the underlying pathophysiological mechanisms and clinical implications.
Key words: Pheochromocytoma; Neurofibromatosis; Arterial hypertension; Treatment
El feocromocitoma es un tumor del sistema simpáticoadrenal, desarrollado a partir de células cromafines, secretoras de catecolaminas, que producen hipertensión arterial secundaria potencialmente curable. Es un tumor infrecuente (1 en 10.000 sujetos) con localización intraabdominal en el 95% de los casos y en el 90% en la médula suprarrenal derecha1.
El feocromocitoma familiar (5%) se hereda de forma autonómica dominante y puede asociarse a neoplasias endocrinas múltiples1.
Las neurofibromatosis son un conjunto heterogéneo de trastornos hereditarios con transmisión también autonómica dominante, con penetrancia completa y variable expresividad clínica, caracterizados por la presencia de anomalías progresivas localizadas en piel, sistema nervioso central; sistema nervioso periférico, esqueleto y glándulas de secreción interna2.
La neurofibromatosis de von Recklinghausen-1 (NF1)3. afecta a 1 de cada 3.000 nacidos de ambos sexos y en el 50% de los casos surge en forma esporádica como una neomutación. El gen de la NF1 se localiza en el cromosoma 17q11.2. Es una afección subdiagnosticada en niños (formas subclínicas u oligosintomáticas). Los signos clínicos más frecuentes en adultos son: presencia de manchas color café con leche, neurofibromas cutáneos, escoliosis y macroencefalia3.
El feocromocitoma puede asociarse a otras enfermedades como la de von Hippel-Lindau4, acromegalia5, dermatomiositis6 y enfermedad de von Recklinghausen7.
Caso clínico
Mujer de 36 años de edad, argentina, soltera, empleada doméstica, que ingresa al servicio de guardia por presentar dolor precordial opresivo, con irradiación a epigastrio, acompañado de disnea, náuseas, cefalea, palpitaciones e hipertensión arterial. La paciente relata cuadros similares desde hace aproximadamente un año tratados en forma sintomática. No refiere hábitos tóxicos, antecedentes personales ni familiares de importancia. En el año 2001, se realizó biopsia de un nódulo en antebrazo derecho que certificó el diagnóstico de neurofibromatosis tipo I.
Al examen físico se constata paciente lúcida, ubicada témporo-espacialmente, con palidez cutánea generalizada, sudoración profusa y temblor de manos. En piel se observan múltiples manchas de color café con leche, de diferentes tamaños en región anterior del tórax, moteado axilar, y un único neurofibroma de 3 mm localizado en región de aréola mamaria derecha, de consistencia blanda, de color marrón, con pedículo móvil.
Esta sintomatología se asocia con presión arterial de 180/ 100 mm Hg, pulso irregular con una frecuencia de 120 latidos por minuto, y frecuencia respiratoria de 24 por minuto.
El laboratorio de rutina, las enzimas cardíacas, incluida la troponina, y la radiografía de tórax fueron normales. El ECG presentó trastornos de repolarización en DII y aVF. El fondo de ojo mostró formaciones nodulares en el iris de ambos ojos que por sus características corresponden a nódulos de Lisch. La perfusión miocárdica con isonitrilo marcado con Tc99 fue normal. El ecodoppler cardíaco constató hipertrofia concéntrica de ventrículo izquierdo y disfunción diastólica leve. El Holter electrocardiográfico mostró extrasistolia supraventricular, con salvas autolimitadas de taquicardia supraventricular.
La resonancia magnética nuclear (RMN) encefálica constato hipoplasia anteromedial temporal izquierda.
La ecografía abdominal mostró en el riñón derecho, en contacto con la valva anterior del polo superior, una imagen hipoecoica de contornos definidos de 28 . 19 mm (Fig. 1).
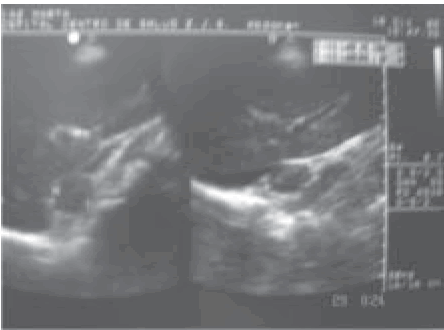
Fig. 1.- Ecografía abdominal: Imagen hipoecoica de 28 . 19 mm en contacto con el polo superior del riñón derecho.
En la tomografía axial computada de abdomen con contraste se observó imagen nodular de 21 . 14 mm de diámetro, bien definida y de bordes netos en la glándula suprarrenal derecha, que desplazaba a la vena cava inferior.
La RMN de abdomen (Fig. 2) reconoció en topografía de la glándula suprarrenal derecha una lesión nodular de 2.5 . 1.8 . 2.3 mm con contornos definidos y parcialmente homogéneos sin características semiológicas de malignidad.
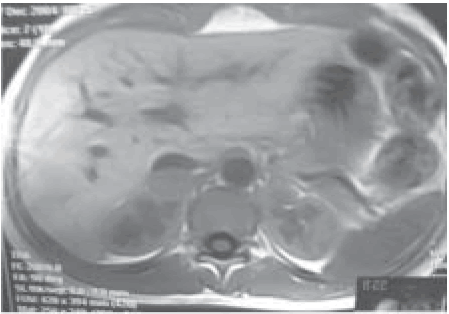
Fig. 2.- Resonancia magnética nuclear: Lesión nodular heterogénea de 2.5 . 1.8 . 2.3 mm con contornos definidos, en topografía de la glándula suprarrenal derecha, desplazando a la vena cava inferior.
Se realizaron determinaciones en orina de ácido vainillinmandélico: 15.6 mg/24 h (VN: hasta 12 mg/24 h), epinefrina: 29.20 µg/24 h (VN: menos de 20 µg/24h) y norepinefrina: 83.30 µg /24 h (VN menos de 90 µg/24 h).
La cámara gamma de cuerpo entero con metaiodobenzilguanidina (MIBG) marcada con I131, sólo mostró captación aumentada del fármaco en la topografía de la glándula suprarrenal derecha.
Con el diagnóstico probable de feocromocitoma, se consideró factible su extirpación quirúrgica por vía laparoscópica. Se realizó tratamiento pre-quirúrgico con bloqueo de receptores α y β, con prasozín y propanolol durante 10 días. Se llevó a cabo exitosamente suprarrenalectomía derecha por vía laparoscópica transabdominal.
La anatomía patológica confirmó el diagnóstico de feocromocitoma con características de benignidad. La paciente evolucionó favorablemente en el post-operatorio y no presentó hipertensión arterial ni crisis hipertensivas en un seguimiento a 36 meses.
La neurofibromatosis tipo 1 evolucionó sin presentar complicaciones que requirieran intervenciones terapéuticas.
Discusión
El diagnóstico de feocromocitoma es fundamentalmente de sospecha, ya que tiene un variado espectro clínico. La signosintomatología más frecuente es la crisis de hipertensión arterial atribuible a hemorragias intratumorales que produce liberación de catecolaminas. Sin embargo, pueden existir feocromocitomas con hipertensión arterial sostenida, hipertensión arterial sostenida con crisis, normo e hipotensión arterial8. En las tres primeras formas clínicas, orienta el diagnóstico la presencia de hipotensión ortostática debido a la secreción por el tumor de adrenomedulina, potente péptido vasodilatador arterial9. En otras situaciones, se asocia con la secreción de péptidos vasoactivos como dopamina, somatostatina, adrenomedulina y péptido intestinal vasoactivo que producen cuadros clínicos de difícil diagnóstico2.
La asociación de feocromocitoma con enfermedad de von Recklinghausen es infrecuente, con una incidencia menor del 1%7. La hipertensión arterial en la enfermedad de von Recklinghausen puede también deberse a estenosis de las arterias renales o ser idiopática2. La neurofibromatosis de von Recklinghausen tiene criterios diagnósticos establecidos por consenso10. Actualmente, se han descripto 8 tipos según la afectación orgánica que produzcan2. Dentro de ellos, exceptuando la NF1, la más característica es la neurofibromatosis tipo 2 o central que se caracteriza por la presencia de neurinoma bilateral del acústico, con escasos o nulos estigmas cutáneos11.
Las neurofibromatosis, a pesar de tener agregación familiar, presentan dentro de una misma familia una expresión fenotípica y gravedad variable12.
En la NF1 el gen responsable produce neurofibromina que es expresada en las neuronas, células de Schwann, médula suprarrenal y leucocitos. Se trata de una proteína ras-GTF (ras-guanidin trifosfato) con propiedades regulatorias debido a una GTF-asa que es capaz de regular la actividad biológica de otras proteínas codificadas por proto-oncogenes de la familia ras12.
Por lo tanto, el gen de la NF1 tendría actividad de supresión tumoral y su mutación o deleción ocasionaría disminución de la actividad GTF-asa con la consiguiente proliferación celular mediada por ras-GTF12.
Ante el diagnóstico de certeza de feocromocitoma se debe intentar su extirpación ya que produce hipertensión arterial potencialmente curable. Se deben tomar recaudos precisos pre, intra y post-operatorios para evitar complicaciones, ya que en el 90% de los casos el tratamiento quirúrgico es curativo1.
Actualmente es una alternativa válida la cirugía laparoscópica, por ser una técnica segura y efectiva en tumores adrenales benignos con pocas complicaciones, que reduce el tiempo operatorio y los días de hospitalización13.
Bibliografía
1. Manger WM, Gifford RW Jr. Pheochromocytoma: a clinical overview In: Laragh JH, Brenner BM (eds.) Hypertension: Pathophysiology, Diagnosis and Management. 2nd ed. New Jork, NY: Raven Press Ltd; 1995: 2225-44.
2. Botto LD, Moore CA, Khoury MJ, Erickson JD. Neural tube defects. N Engl J Med 1999; 341: 1509-19.
3. Gutman D. Neurofibromatosis-1. Neurol Clin 2002; 841-65.
4. Touiti D, Seket B, Deligne E, et al. Bilateral adrenal pheochromocytomas in Von Hippel-Lindau disease. Ann Urol 2001; 35: 323-328.
5. Sleilati GG, Kovacs KT, Honasoge M. Acromegaly and pheochromocytoma: report of a rare coexistence. Endocr Pract 2002; 8: 54-60.
6. Yeh CN, Jeng LB, Chen MF, et al. Nonfunctioning malignant pheochromocytoma associated with dermatomyositis: case report and literature review. World J Urol 2001; 19: 148-150.
7. Takayama T, Kato Y, Tsuru N, et al. A case of pheochromocytoma with Von Recklinghausen's and review of 67 japanese cases. Nippon Hinyokika Gakkai Zasshi 2001; 92: 479-83.
8. Bravo E, Guifford R, Manger W. Adrenal medullary tumors: Pheochromocytoma. In Mazzaferri E, Samaan N. (eds). Endocrine tumors. Boston, Blackwell Scientific Publications 1993; 426-47.
9. Samson W. A novel vascular hormona: adrenomedullin. In: Sowers J, (ed). Endocrinology of the vasculature. Totowa N: Humana Press 1997; 269-81.
10. National Institutes of Health Consensus Development Conference. Neurofibromatosis. Conference Statement. Arch. Neurol 1988; 45: 575-8.
11. Baser M, Evans D, Gutmann D. Neurofibromatosis 2. Curr Opin Neurol 2003; 16: 27-33.
12. Gutmann DH, Collins FS. Recent progress toward understanding the molecular biology of von Reckling- hausenneurofibromatosis. Ann Neurol 1992; 31: 555-61.
13. Kuriansky J, Shabtai M, Ayalon A. Laparoscopic adrenalectomy for pheochromocytoma. Harefuah 2001; 140: 214-16. [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ]
Recibido: 23-02-2007
Aceptado: 4-07-2007

