










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.67 n.6-2 Ciudad Autónoma de Buenos Aires nov./dic. 2007
La ADN topoisomerasa tipo I de protozoos patógenos como Diana terapéutica de fármacos antitumorales
Rosa M. Reguera, Yolanda Pérez-Pertejo, Cármen M. Redondo, Rosario Díaz-González, Rafael Balaña-Fouce
Departamento de Farmacología y Toxicología (INTOXCAL), Universidad de León, España
Dirección postal: Dr. Rafael Balaña Fouce, Dpto. Farmacología y Toxicología, Universidad de León, Campus de Vegazana s/n, 24071 León, España Fax: (00-34) 987291252 e-mail: rbalf@unileon.es
Resumen
La utilización intensiva de fármacos antiparasitarios es la causa principal de la aparición de microorganismos parásitos multirresistentes en las regiones del planeta donde son precisamente endémicos. Los agentes etiológicos de las denominadas enfermedades tropicales -malaria, criptosporiodiosis, enfermedad del sueño, enfermedad de Chagas o los distintos tipos de leishmaniosis- son protozoos unicelulares sobre los que no se ha desarrollado en la actualidad ninguna vacuna eficaz y cuyo tratamiento se basa en medidas sanitarias preventivas y en el uso de medicamentos. La quimioterapia antiparasitaria actual es cara, no está ausente de efectos adversos y no supone beneficios a las empresas que la comercializan, por lo que la inversión en I & D es marginal comparada con la llevada a cabo para otros procesos patológicos de menor relevancia médica. La identificación de las ADN topoisomerasas como dianas farmacológicas se basa en los excelentes resultados obtenidos en los ensayos clínicos llevados a cabo con los derivados de la camptotecina en la terapia antitumoral. Las importantes diferencias estructurales entre las ADN topoisomerasas de tipo I de tripanosomas y leishmanias con respecto a sus homólogas de mamífero ha abierto un nuevo campo de investigación que combina las técnicas de biología molecular con la cristalización de proteínas para poder diseñar nuevos fármacos dirigidos específicamente a su inhibición. Revisamos aquí las características de estas nuevas dianas farmacológicas, así como los compuestos que en el momento están siendo utilizados para su inhibición en los agentes parasitarios que causan las principales enfermedades tropicales.
Palabras clave: Topología del ADN; ADN topoisomerasa tipo I; Camptotecina; Enfermedades tropicales
Abstract
Type I DNA topoisomerase from protozoan pathogens as a potential target for anti-tumoral drugs. The intensive use of antiparasitic drugs is the main cause of the emergence of multiresistant parasite strains on those regions where these parasites are endemic. The aetiological agents of the so-called tropical diseases viz. malaria, cryptosporidiosis, sleeping sickness, Chagas disease or leishmaniasis, among others, are unicellular protozoan parasites with no immune-prophylactic treatment and where the chemotherapeutical treatment is still under controversy. At present, the chemotherapeutic approach to these diseases is expensive, has side or toxic effects and it does not provide economic profits to the Pharmaceuticals which then have no or scarce enthusiasm in R & D investments in this field. The identification of type I DNAtopoisomerases as promising drug targets is based on the excellent results obtained with camptothecin derivatives in anticancer therapy. The recent finding of significant structural differences between human type I DNAtopoisomerase and their counterparts in trypanosomatids has open a new field in drug discovery, the aim is to find structural insights to be targeted by new drugs. This review is an update of DNA-topoisomerases as potential chemotherapeutic targets against the most important protozoan agents of medical interest.
Key words: DNA topology; Type I DNA topoisomerase; Camptothecin; Tropical diseases
ABREVIATURAS:
TOPI: ADN-topoisomerasa tipo I
TOPII: ADN-topoisomerasa tipo II
hTOPI: ADN-topoisomerasa tipo I humana
LdTOPI: ADN-topoisomerasa tipo I de Leishmania donovani
TbTOPI: ADN-topoisomerasa tipo I de Trypanosoma brucei
PfTOPI: ADN-topoisomerasa tipo I de Plasmodium falciparum
NLS: señal de localización nuclear
ADNk: ADN de kinetoplasto
ROS: especies reactivas de oxígeno
Lk: número de enlace
CPT: camptotecina
REB: rebecamicina
QSAR:relación estructuraactividad
Tdp-I: tirosil-ADN-difosfodiesterasa-1
SUMO:modificadores tipo ubiquitina de pequeño tamaño
Las enfermedades tropicales producidas por parásitos unicelulares se encuentran entre las plagas más dañinas que asolan el planeta debido a las enormes pérdidas en vidas humanas y a los elevadísimos costes económicos resultantes de su morbilidad. Malaria, criptosporidiosis, enfermedad del sueño, enfermedad de Chagas, o leishmaniosis, entre otras, son enfermedades que tienen un gran impacto en el estado sanitario de grandes masas poblacionales que viven al límite de la pobreza.
La malaria o paludismo constituye un problema sanitario de gran envergadura, ya que más del 40% de la población mundial es susceptible de padecerla actualmente, siendo la segunda enfermedad de declaración obligatoria, después de la tuberculosis, que causa mayor índice de mortalidad en humanos. La malaria está causada por protozoos unicelulares del género Plasmodium (P. falciparum y P. vivax son los de mayor importancia epidemiológica) trasmitidos al ser humano por la picadura de mosquitos del género Anopheles que previamente han estado en contacto con un individuo infectado. Dentro del hospedador humano, el parásito experimenta una serie de cambios como parte de su complejo ciclo vital que producen fiebres intermitentes y anemia, que pueden causar la muerte del individuo. La malaria es endémica en más de 100 países, la mayoría situados en las regiones tropicales de África y Asia, donde las medidas de control y las condiciones sanitarias son claramente insuficientes1.
La familia Trypanosomatidae (tripanosomas y leishmanias entre otros) tiene como rasgo característico en común la presencia de un flagelo insertado en su única mitocondria que alberga un orgánulo especializado conocido como kinetoplasto. El kinetoplasto contiene miles de cadenas circulares de ADN (ADNk) que se enredan en marañas conocidas como maxi- y minicírculos que por su peculiar organización han atraído la curiosidad de biólogos moleculares para desentrañar su función, y de los que hablaremos más adelante2. Los tripanosomas africanos causan la enfermedad del sueño que es transmitida por picaduras de la mosca tse-tse. Los agentes etiológicos de esta enfermedad son el Trypanosoma brucei gambiense, en los países del Africa Central y Occidental, que causa un tipo de infección crónica que puede ser asintomática durante meses o incluso años; y el T. b. rhodesiense, que por el contrario origina brotes repentinos, agudos y virulentos, en los países del Este y Sur del África Subsahariana3.
Trypanosoma cruzi es el agente etiológico de la enfermedad de Chagas en países del Centro y Sur de América. Los seres humanos, que se comportan como hospedadores definitivos, contraen la enfermedad por las picaduras de un insecto hemíptero de la sub-familia Triatominae, conocido como chinchorro o vinchuca según el país de origen. Tras su inoculación en el individuo, el parásito invade el torrente sanguíneo y se multiplica en el interior de las células de la musculatura cardíaca y lisa originando cardiomiopatías graves y megaformaciones del tracto digestivo4.
Por el término leishmaniosis se conoce a un conjunto de enfermedades zoonóticas transmitidas por las hembras de los mosquitos de los géneros Phlebotomus y Lutzomyia que originan afecciones cutáneas, mucocutáneas y viscerales en el hombre y otros animales. La leishmaniosis visceral humana -kala azar- es producida por L. donovani, un microorganismo unicelular que parasita los macrófagos del sistema retículo-endotelial del hospedador. Es la forma más dañina de estas enfermedades, produciendo fiebre, espleno-hepatomegalia y anemia, pudiendo ser fatal si no se diagnostica y se trata a tiempo. Cerca del 90% de los casos de leishmaniosis visceral tienen su origen en India, Bangladesh, Indonesia y Sudan5. La leishmaniosis es también importante en América Central y del Sur, en este caso por formas endémicas que originan afecciones mucocutáneas de la enfermedad, más leves pero muy insidiosas y deformantes.
La ciencia no ha desarrollado aún el remedio universal frente a las enfermedades tropicales y es muy poco probable que una solución única exista alguna vez. Se sabe desde hace mucho tiempo que el estado inmunológico del hospedador es fundamental en la cura y en el desarrollo de inmunidad adquirida de estas enfermedades. Sin embargo, a pesar de los esfuerzos empleados aún no existe una vacuna eficaz para evitar ninguna de estas dolencias.
El tratamiento de la malaria se ha basado en fármacos como la cloroquina, mefloquina, primaquina y quinina, pero su uso intensivo durante décadas ha producido formas multirresistentes del parásito en casi todos los países endémicos6. Los tratamientos basados en la combinación de fármacos, como el cocktail de pirimetaminasulfadoxina7 o los que tienen como base fármacos de origen natural como la artemisinina y sus combinaciones (por ejemplo; artesunato-mefloquina) están sustituyendo a las terapias clásicas tanto en Africa como en Asia8.
La quimioterapia desarrollada frente a la enfermedad del sueño no carece de efectos adversos y en muchos lugares han aparecido cepas resistentes que han reducido drásticamente su eficacia. Los fármacos de elección; pentamidina -una diamidina aromática- y la suramina -una naftilamina sulfonada- aún tienen utilidad frente a los estadios tempranos de la enfermedad9. Frente al segundo estadio, en el que se ve afectado el SNC, se utilizan tres fármacos de elección; el melarsoprol -un derivado arsenical-, el inhibidor irreversible de la síntesis de poliaminas, eflornitina, y el nifurtimox10,11.
La farmacopea existente frente a la enfermedad de Chagas incluye un derivado nitroimidazólico -el benznidazol- y el nifurtimox -un derivado del nitrofurano- que actúan mediante la generación de radicales libres de oxígeno (ROS) destruyendo con cierta selectividad las células del parásito debido a su menor capacidad antioxidante12, 13. Tanto el nifurtimox como el benznidazol tienen una actividad terapéutica significativa en las fases agudas de la enfermedad pero no en las crónicas donde el tratamiento falla por completo.
El arsenal terapéutico utilizado frente a los distintos tipos de leishmaniosis incluye a los antimoniales pentavalentes (Pentostam® y Glucantime®, de los que hablaremos más adelante), el antibiótico poliénico anfotericina B y la pentamidina14, 15. Recientemente, la incorporación del derivado de la alquilfosfocolina (miltefosina: Miltex®) ha supuesto un avance significativo en el tratamiento de las formas viscerales de esta enfermedad16.
Sumados a estos inconvenientes hay que señalar que la mayoría de estos fármacos no son fáciles de manejar, requieren tratamientos largos y costosos y en muchas ocasiones no están libres de efectos colaterales indeseables. Por otro lado, la sombra de estas enfermedades amenaza hoy por hoy a los habitantes de los llamados países desarrollados. Los intercambios culturales y turísticos, fruto de la globalización del planeta y los procesos de inmunodepresión ligados a ciertas enfermedades, han hecho renacer el fantasma de estas dolencias allí donde se pensaba que habían sido definitivamente erradicadas17-19. Es por esto que el diseño y desarrollo de fármacos frente a estas enfermedades suscita un creciente interés en la comunidad científica internacional, ocupando el lugar dejado por las grandes empresas farmacéuticas que no encuentran estímulo en hacer inversiones en I + D allí donde no se esperan grandes beneficios económicos.
La topología del ADN
En 1965 Vinogard y col.20 obtuvieron las primeras microfotografías electrónicas de ADN circular del virus del polioma (SV40). Bajo ciertas condiciones experimentales la molécula de ADN adquiría una conformación retorcida muy compacta que recordaba la estructura de una goma circular elástica girada varias veces sobre sí misma. Esta forma topológica del ADN recibió el nombre de superenrollada ("supercoiled") en oposición a la forma topológica no retorcida a la que se denominó relajada ("relaxed"). El superenrollamiento de una molécula circular de ADN se produce por la torsión generada por un exceso, o por un defecto, en el número de enlace (Lk) entre las dos cadenas antiparalelas de la doble hélice. En un estado de superenrollamiento la información codificada en el material genético no es accesible a los mecanismos de replicación y transcripción, por lo que esta torsión debe disiparse reduciendo el valor Lk a 0.
Para relajar el ADN superenrollado deben producirse cortes en las cadenas de ADN circulares que permitan rotar libremente a sus extremos. Por su parte, las moléculas de ADN lineales necesitan relajar dominios discretos entre estructuras que se mantienen condensadas. Los mecanismos de relajación del ADN superenrollado deben operar cortando una o las dos cadenas del ADN para que las atraviesen otras cadenas, seguido del sellado de las mismas, con el fin de restaurar la continuidad e integridad del mensaje genético. Estos cambios en la topología del ADN se producen enzi-máticamente mediante proteínas que originan cortes transitorios en el ADN y permiten el paso de la otra hebra por el hueco creado antes de su sellado.
ADN topoisomerasas
Las ADN topoisomerasas catalizan cambios en la topología del ADN durante los procesos de replicación, transcripción, recombinación y reparación del genoma. En primer lugar, pueden cortar y empalmar repetidamente los enlaces fosfodiéster del esqueleto de polifosfodesoxiribosa que alberga en su interior a las bases nitrogenadas codificadoras del mensaje genético. En segundo lugar, permiten que otras cadenas de ADN pasen entre los dos cabos transitoriamente escindidos. En el desempeño de esa tarea, las topoisomerasas utilizan la energía del enlace internucleotídico para unirse covalentemente al extremo 3' o 5' del ADN. Cuando empalman nuevamente las cadenas de ADN, revierten esa unión covalente, restableciendo el enlace internucleotídico inicial21-24.
Se han caracterizado tres tipos de ADN topoisomerasas en función de sus propiedades catalíticas, gasto energético y estructura de su proteína: las ADN topoisomerasas de tipo I (TOPI, subtipos IA y IB) y las ADN topoisomerasas de tipo II (TOPII).
Las TOPII son enzimas homodiméricas que generan cambios topológicos complejos del ADN (relajación, desanudado y desencadenado) mediante la ruptura transitoria de las dos cadenas de la doble hélice y gasto energético en forma de ATP. Durante el proceso de ruptura y empalme, se produce un intermediario covalente entre los extremos 5' de cada una de las hebras del ADN y cada una de las subunidades enzimáticas. Se han identificado y expresado funcionalmente las de varios parásitos como: P. falciparum25, Cryptosporidium parvum26, T. brucei27, T. cruzi28, y varias especies del género Leishmania 29,30, sin que existan grandes diferencias morfológicas o cinéticas con las de sus correspondientes hospedadores.
Al contrario de lo que sucede con las TOPII, las enzimas de tipo I rompen sólo una de las hebras del ADN y únicamente permiten relajar el ADN superenrollado. Se subdividen en función de que formen el intermediario covalente con el extremo 5' del segmento escindido (IA) o con el extremo 3' (IB). Este intermediario se establece tras el ataque nucleofílico del grupo hidroxilo de una tirosina presente en su centro activo con un grupo fosfato de la cadena nucleotídica, originando un enlace fosfodiéster transitorio que rompe la cadena del ADN. En este momento la hebra mellada gira a través de su complementaria alcanzándose un mayor grado de relajación31. Finalmente, la enzima religa la cadena mellada. Las TOPI son enzimas monoméricas -con la excepción que va mos a comentar más adelante- y no necesitan ATP para relajar el ADN superenrollado positiva o negativamente (Fig. 1).

Fig. 1.- A. Las ADN-topoisomerasas de tipo I catalizan la relajación del ADN superenrollado circular in vitro. B. La fotografía muestra un ensayo convencional para evaluar la actividad TOPI de una muestra biológica. Para ello se incluyen muestras de ADN relajado (carril 1) y superenrollado (carril 2) que sirven como patrones de la reacción. El ADN superenrollado se incluye como sustrato de la reacción que a diferentes concentraciones de enzima o de tiempos, irá relajándose gradualmente apareciendo diferentes topoisómeros. La imagen muestra la comparación entre la enzima recombinante humana (hTOPI) y la de L. donovani (LdTOPI) y procede de una caracterización rutinaria realizada por los autores.
La TOP I humana (hTOPI) ha sido caracterizada estructuralmente con gran detalle y sirve como modelo comparativo para el resto de las enzimas homólogas de otras procedencias. La hTOPI es una enzima monomérica de 765 aminoácidos con una masa molecular teórica de 91 kDa. Los estudios cristalográficos permiten distinguir cuatro dominios estructurales: i) el extremo N-terminal, cargado positivamente y poco conservado filogenéticamente. Contiene cuatro posibles señales de localización nuclear (NLS) y no es necesario para la actividad de relajación; ii) el dominio central ("core") es esencial para relajar el ADN superenrollado y muestra un alto grado de conservación filogenética; iii) el extremo C-terminal contiene el residuo de Tyr-723 que establece el enlace fosfodiéster transitorio con el ADN, y iv) conectando el dominio C-terminal con la región central "core" se encuentra una región de escasa conservación filogenética denominada "linker" que no interviene funcionalmente en la relajación del ADN32-34.
La TOPI de protozoos parásitos
La TOPI del parásito de la malaria P. falciparum fue descrita por primera vez por Riou y col.35 en 1986, quienes la purificaron y caracterizaron a partir de eritrocitos infectados. Es una enzima monomérica de 839 aminoácidos y 104 kDa de masa molecular. El gen que codifica para esta proteína (PfTopI) aparece como copia única en el cromosoma 5 del genoma del parásito36. Estructuralmente la proteína guarda un 42% de homología con la enzima humana, con una composición estructural muy parecida a la de su hospedador: el extremo N-terminal, poco conservado y de 134 aminoácidos, un bloque de 500 aminoácidos que tienen alta homología con el dominio "core" de la proteína humana y el extremo C-terminal, más pequeño que el de la enzima del hospedador pero que contiene la tirosina del centro activo. La expresión de la ADN topoisomerasa I en P. falciparum (PfTOPI) difiere en los diferentes momentos de su ciclo celular37. De este modo se encuentran altos niveles de ARNm PfTOPI en el estado de trofozoito pero no en el de esquizonte.
Las TOPI de tripanosomas y leishmanias difieren significativamente del resto de las enzimas homólogas descritas hasta el momento. Como dijimos anteriormente, estas enzimas son monoméricas en todas las especies animales, vegetales y de otros microorganismos. Sin embargo, durante el proceso de caracterización genómica de las TOPI de L. infantum y L. donovani (LdTOPI), se encontró sorprendentemente que éstas presentaban una forma truncada que carecía de la secuencia codificante para el centro activo. La búsqueda de la región perdida dentro del genoma de Leishmania dio como resultado la aparición de un segundo gen que contenía todos los motivos complementarios de los que carecía el primer gen descubierto. En conclusión, la LdTOPI esta formada por dos subunidades, una de 636 aminoácidos (masa molecular teórica: 73 kDa) codificada por el gen LdTOPIA, situado en el cromosoma 34, y otra de 262 aminoácidos con una masa molecular teórica de 28 kDa, codificada por el gen LdTOPIB que se encuentra en el cromosoma 438.
Más tarde Bodley y col.39 observaron en 2003 que esta constitución dimérica ocurría también en T. brucei, y el análisis genómico confirmaba que la existencia de dos genes independientes codificando para las regiones "core" (TbTOPIA) y catalítica (TbTOPIB) era un hecho común para todos los tripanosomátidos.
Comparando la secuencia de aminoácidos de la TOPI heterodimérica de tripanosomátidos con las de otros organismos eucariotas encontramos algunas diferencias destacables: El extremo N- terminal de LdTOPIA es corto y poco conservado comparado con el extremo amino terminal de otras TOPI. A esta región le sigue el dominio central "core", el cual sí muestra una alta homología con el resto de enzimas, y en la que podemos encontrar la mayor parte de los aminoácidos que interaccionan con el ADN. Finalmente el extremo C-terminal carece de cualquier homología con otras TOPI 38. Por su parte, LdTOPIB contiene un extremo N-terminal extenso rico en serinas, susceptibles de fosforilación (posible mecanismo de regulación post-traduccional). Su extremo carboxilo guarda homología con el dominio C-terminal del resto de TOPI y en él se encuentra la Tyr-222 necesaria para la ruptura de la cadena nucleotídica del ADN (Fig. 2).

Fig. 2.- Representación esquemática lineal de la TOPI humana (hTOPI) comparada con la de protozoos parásitos representativos. Los números de acceso al banco de datos GeneBank son los siguientes: Homo sapiens (hTOPI), K03077; P. falciparum (PfTOPI), Q26024; C. parvum (CpTOPI), Q5CY81; L. donovani subunidad grande (LdTOPIA) AF303577; L. donovani subunidad pequeña (LdTOPIB) AY062908; T. brucei subunidad grande (TbTOPIA) Q581U8 y T. brucei subunidad pequeña (TbTOPIB) AAP78905. Los estudios estructurales con la enzima humana, dividen a esta proteína en cuatro dominios estructurales, que por analogía de secuencia pueden extenderse a las otras especies: i) dominio hidrofílico N-terminal no conservado; ii) dominio central ("core") hidrofóbico de interacción con el ADN muy conservado; iii) dominio de unión ("linker") que conecta el "core" con el centro activo, y iv) dominio C-terminal que contiene el centro activo implicado en cortar transitoriamente una de las cadenas de ADN. A diferencia de las especies del orden Apicomplexa, las TOPI de tripanosomátidos contienen los residuos catalíticos compartidos entre las dos subunidades.
La localización del dominio "linker" en la enzima dimérica de tripanosomátidos es hasta el momento desconocida. Podría estar formado por parte de las dos subunidades que al interaccionar estabilizaran el conjunto, o bien podría generarse tras modificaciones posttraduccionales que eliminaran parte de los extremos amino o carboxilo de las diferentes subunidades (Fig. 3). Estudios realizados con la enzima de Leishmania en los que se ha deleccionado parte de los extremos N- y Cterminales de ambas subunidades muestran que casi 70 aminoácidos del extremo carboxilo de la LdTOPIA no son necesarios para la actividad catalítica, aunque pueden contener posibles NLS; del mismo modo el extremo amino de la LdTOPIB no es necesario para la actividad catalítica (Balaña-Fouce y col. datos inéditos).
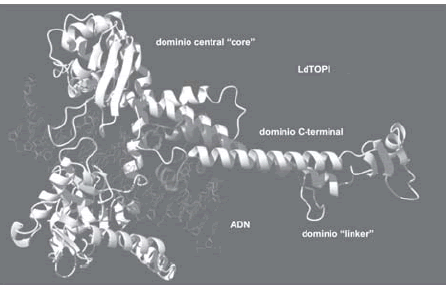
Fig. 3.- Vista tridimensional perpendicular de la TOPI dimérica del tripanosomátido L. donovani (LdTOPI) junto con el ADN. Se observa el dominio central "core" así como el dominio de conexión o "linker" y el extremo C-terminal, ya perteneciente a la subunidad pequeña. LdTOPI ha sido simulada utilizando el programa Swiss pdb por analogía con la enzima humana (hTOPI); los dominios se han establecido por comparación con los descritos por Redinbo y col.34.
Las TOPI de tripanosom átidos se encuentran tanto en núcleo como en kinetoplasto40. Dado que la actividad de desencadenado y desanudado del ADNk se atribuye a la TOPII, se desconoce la función de la enzima tipo I en este orgánulo. La anulación génica de la TOPII de tripanosomátidos produce la pérdida del kinetoplasto (diskinetoplasia), una condición que conlleva la muerte del parásito41. Por otra parte, el silenciamiento de cualquiera de las dos subunidades de la TOPI de T. brucei (TbTOPI) mediante técnicas de ARN de interferencia origina la pérdida de viabilidad de los tripomastigotes in vitro lo que indica la necesidad de ambas actividades enzimáticas para la supervivencia de estos microorganismos parásitos 42.
La TOPI como diana terapéutica de fármacos antitumorales
Desde su descubrimiento las TOPI han atraído poderosamente la atención de la comunidad científica como potenciales dianas de intervención terapéutica, debido a su posición estratégica en la replicación, transcripción y recombinación del material genético. Debido a este papel clave en el mantenimiento de la fidelidad de la información genética, su estructura se ha mantenido conservada filogenéticamente lo que hace que cualquier fármaco diseñado para su inhibición no discrimine entre la enzima del parásito invasor y la del hospedador, suponiendo un alto riesgo de toxicidad añadido en detrimento de su efecto terapéutico. Sin embargo, el reciente descubrimiento de una TOPI en tripanosomátidos que difiere estructuralmente de la del hospedador, así como su diferente grado de expresión durante los procesos de crecimiento rápido, ha abierto una nueva ventana al diseño de moléculas que actúen específicamente sobre la forma del parásito43-45.
Clásicamente los inhibidores de las TOPI se han clasificado en dos categorías: i) los compuestos que estabilizan el complejo de escisión entre la enzima y el ADN se han denominado genéricamente inhibidores de clase I o venenos enzimáticos; ii) por su parte los compuestos que interfieren las funciones catalíticas de la enzima se han denominado inhibidores de clase II46, 47 (Fig. 4).

Fig. 4.- Estructura química de algunos inhibidores de la TOPI: (A) CPT; (B) compuestos heterocíclicos distintos a la CPT; (C) compuestos que se unen al surco menor de la hélice de ADN.
La camptotecina (CPT) y sus derivados son el mejor ejemplo de un veneno de clase I. Estructuralmente la CPT es un alcaloide natural pentacíclico (Fig. 4A) producido por la planta de la familia de las Nisáceas Camptotheca accuminata, originaria de China y el Tibet. Esta molécula lidera una serie enorme de compuestos con actividad antitumoral que en la actualidad están integrándose en la quimioterapia de ciertos tipos de cánceres48-50. Bioquímicamente la CPT es un potente inhibidor no competitivo de la TOPI con la que establece un complejo ternario irreversible junto con el ADN mellado (complejo de escisión) que no puede ser sellado. Como consecuencia se producen mellas en aquellas zonas del genoma que estén replicándose en ese momento, lo que provoca la muerte de la célula si no se induce la reparación de las mismas.
Este mecanismo de acción de la CPT ha sido comprobado en protozoos parásitos de interés médico51. Bodley y col.52 demostraron en 1998 que la CPT induce la formación de complejos de escisión en el interior del núcleo de las formas intraeritrocitarias de P. falciparum lo que origina la muerte del parásito a concentraciones micromolares del fármaco. Los mismos autores realizaron estudios de relación estructura/actividad (QSAR) una serie de análogos de la CPT sobre tripomastigotes de T. brucei, demostrando que su citotoxicidad se correlacionaba con la capacidad de formación de complejos de escisión con el ADN y la enzima. Estudios más recientes han revelado que los derivados hidrosolubles de la CPT, irinotecan y topotecan, que se utilizan clínicamente como fármacos antitumorales, tienen un efecto citotóxico reducido frente a las formas sanguíneas de T. brucei53. Los autores han concluido que la reducida eficacia de estos compuestos se debe más a su baja permeabilidad para atravesar la membrana del parásito que a su eficacia de formar complejos de escisión.
Basado en el análisis de los cristales de la hTOPI junto con ADN y CPT, Staker y col.54,55 han propuesto en 2005 un modelo de interacción entre los diferentes componentes del complejo de escisión que podría ser asumido en tripanosomátidos (teniendo en cuenta su estructura dimérica). La Fig. 5 muestra este modelo de interacción utilizando la LdTOPI que es la que han caracterizado los autores de la presente revisión.

Fig. 5.- Interacciones de la CPT con algunos aminoácidos de la TOPI de L. donovani (LdTOPI). Los enlaces por puentes de hidrógeno se muestran en líneas discontinuas. Los aminoácidos que pertenecen a la subunidad grande son Arg 190, Lys 352 y Asp 353; mientras que los pertenecientes a la subunidad pequeña son Asn 221 y Tyr 222.
Derivado de este modelo se ha establecido que la CPT se intercala en el sitio de escisión del ADN mimetizando a una base nitrogenada. Dentro del lugar de intercalación la cadena lateral del Asp-533 de la enzima humana (que se corresponde con el Asp-353 de LdTOPIA) establece un enlace por puente de hidrógeno con el radical 20(S)-hidroxilo de la forma lactónica de la CPT, mientras que la Asn-221 -el aminoácido adyacente a la Tyr-222 del centro activo de la LdTOPIB- crea un segundo puente de hidrógeno con la CPT. La Arg-364, (que se corresponde con la Arg-190 de la LdTOPIA) es igualmente necesaria para la inhibición de la CPT aunque no establece enlaces por puentes de hidrógeno con el fármaco. Otros aminoácidos importantes que interaccionan con la CPT son: Phe-361, Gly-363 y Arg-364 del dominio central situado en LdTOPIA56.
Como otros compuestos que inducen daños en el ADN, los inhibidores de clase I se comportan como inductores de la muerte celular programada57. Sen et al.58 demostraron en 2004 que el efecto leishmanicida inducido por la CPT aparecía inmediatamente después de que su función mitocondrial fuera inhibida, lo que originaba un incremento en su potencial de membrana. Este desacoplamiento iba ligado a un incremento en la concentración de ROS, con el concomitante efecto sobre la peroxidación lipídica de membranas y disminución de la concentración intracelular de glutation reducido. Como en las células de mamífero, se han descrito que otros marcadores apoptóticos como las proteasas de tipo caspasa, la liberación de Ca2+ de los compartimentos intracelulares y la activación de la poli(ADP-ribosa) polimerasa eran igualmente producto de la exposición de los promastigotes de L. donovani a CPT59.
Se han realizado muy pocos estudios para determinar la actividad de la CPT y de sus análogos en infecciones experimentales. En 2001 Proulx et al.60 estudiaron la eficacia de una formulación de CPT encapsulada en liposomas en un modelo murino de leishmaniosis visceral, observando una reducción significativa de la carga parasitaria a la dosis de 2.5 mg/kg peso vivo cuando se administraba por vía intraperitoneal.
Los derivados indolocarbazólicos (rebecamicina; REB y derivados) constituyen un prometedor grupo de inhibidores de clase I de la TOPI con estructura poliheterocíclica producidos naturalmente por el actinomiceto Saccharotrix aerocolonigenes. Muchos derivados semisintéticos de la REB se han ensayado in vitro para evaluar su QSAR frente a multitud de líneas tumorales humanas y de animales, que han servido para comenzar algunos ensayos clínicos de importancia61. La estructura poliheterocíclica plana de estas moléculas les permite intercalarse entre las pares de bases de ADN donde finalmente se establecen los complejos ternarios junto con el ADN y la enzima. En 2005, Deterding y col.62 encontraron que la REB presentaba un buen efecto citotóxico frente a tripomastigotes de T. brucei en concentraciones sub-micromolares. Este efecto fue corroborado por nuestro grupo (Balaña-Fouce y col., resultados inéditos) utilizando una serie de análogos de la REB frente a promastigotes de L. donovani. Nuestros resultados han demostrado que la presencia de un sustituyente azucarado en la molécula de REB incrementa la estabilidad del complejo de intercalación al interaccionar con el surco menor externo de la doble hélice de ADN.
Una tercera clase de inhibidores de clase I de las TOPI son los compuestos con estructura indenoisoquinolínica (Fig. 4B). Igual que los compuestos anteriores estas moléculas se encuentran en fase preclínica como agentes antitumorales63. Nuestro grupo ha evaluado una serie de indenoisoquinolinas frente a promastigotes de L. major in vitro, encontrando un prometedor índice terapéutico con la molécula MNR 2-50 con la que se están realizando ensayos en infecciones experimentales de leishmaniosis visceral (Balaña-Fouce et al., resultados inéditos).
L. donovani es muy sensible a otros fármacos inhibidores de clase I de TOPI como es el derivado naftoquinónico diospirina64. La diospirina es un inhibidor relativamente específico de la enzima de Leishmania, necesitándose concentraciones diez veces superiores para producir un efecto semejante en la enzima homóloga de mamífero65. Por su parte, los derivados del ácido betalínico con estructura de triterpenoides pentacíclicos actúan previniendo la formación del complejo binario enzima- ADN aunque, a diferencia de los compuestos anteriores, no forman parte del mismo66. Este compuesto tiene un efecto inhibitorio drástico sobre el crecimiento de promastigotes de L. donovani en los que inducen la marginación de la cromatina y subsiguiente descromatinación, aunque no se han encontrado efectos sobre el ADNk67.
La berberina es otro inhibidor de clase I de la TOPI con estructura poliheterocíclica semejante al fármaco antitumoral intercalante benzo[a]acridina. La berberina y alguno de sus análogos, han sido estudiados in vitro frente a promastigotes de L. donovani68 y formas sanguíneas de T. brucei y T. congolense69, mostrando eficacia en el rango micromolar. Los ensayos en experimentación animal con estos compuestos han mostrado su eficacia en infecciones experimentales de hámster.
Finalmente, indicaríamos que ciertos compuestos que se han venido utilizando desde hace décadas como antiparasitarios, pueden comportarse como inhibidores de la TOPI en ensayos in vitro. Entre estos inhibidores resaltaríamos los fármacos leishmanicidas de elección derivados del antimonio pentavalente; Glucantime® (antimoniato de meglumina) y Pentostam® (estibogluconato de sodio70, 71). Estos compuestos estabilizan los complejos binarios de escisión in vitro, aunque no parece que constituya su mecanismo de acción principal in vivo72. Por otro lado, los compuestos que se unen al surco menor externo del ADN (Fig. 4C) tales como los derivados bis-benzimidazólicos (Ho-33342, Ho-33258) y las diamidinas aromáticas pentamidina y berenil, pueden inhibir la TOPI in vitro73. Estos compuestos se unen a las regiones ricas en AT del ADN interfiriendo en la catálisis enzimática pero con la excepción del Ho-33342 y del Ho- 33258 no estabilizan los complejos escisión. Un trabajo reciente demuestra sin embargo que el efecto leishmanicida de estos compuestos está relacionado sólo marginalmente con la inhibición de la TOPI, indicando un comportamiento pleiotrópico más complejo74.
TOPI y reparación del material genético
Ha quedado establecida la importancia de las TOPI en el mantenimiento y replicación del material genético; sin embargo por su propia naturaleza, estas enzimas pueden ocasionar daños en el ADN cuando los complejos de escisión se estabilizan por la presencia de inhibidores de clase I. Cuando la maquinaria de transcripción y/o replicación colisiona con los complejos de escisión, se van a producir roturas de cadena sencilla o doble que pueden dar origen a mutaciones puntuales, fragmentación del genoma o muerte celular por apoptosis si no son debidamente reparadas75,76. En células de mamífero se ha descrito una proteína involucrada en la restauración de los complejos de escisión; la tirosil-ADN difosfodiesterasa I (Tdp-1) enzima que cataliza la ruptura del enlace fosfotirosina entre la TOPI y el ADN produciendo un extremo 3' que es procesado a continuación por una polinucleótido kinasa 3'-fosfatasa 77.
Una vez separada del complejo de escisión la TOPI va a ser sustrato de mecanismos de regulación posttraduccionales, bien por: i) degradación proteosomal previo marcado con ubiquitina; o bien por ii) relocalización desde el nucleoplasma al nucléolo mediante el marcado previo con los denominados modificadores tipo ubiquitina de pequeño tamaño (small ubiquitin-like modifiers; SUMO).
La degradación proteosomal previa ubiquitinación, es un mecanismo de regulación post-traduccional específico de la TOPI fosforilada e independiente del estado de replicación de la célula78,79. Pommier80 en 2004 ha propuesto que la degradación por el proteosoma 26S puede ser de utilidad para: i) aumentar la tolerancia a los inhibidores de clase I; ii) facilitar la actividad Tdp1 en la ruptura del enlace fosfotirosina entre la TOPI y el ADN. Varios autores han demostrado que los inhibidores del proteosoma actúan sinergísticamente con la CPT en la inducción de la muerte celular programada en células animales81.
Además de la degradación proteosomal de la TOPI se ha descrito una respuesta transitoria a la CPT que supone el marcado previo de la enzima con modificadores SUMO82. La conjugación con SUMO (SUMOilación) es un proceso independiente de la replicación del cromo- soma que, a diferencia de la ubiquitinación, se produce con la enzima desfosforilada y no persigue la degradación proteínica de la enzima83. El mecanismo de SUMOilación no es un sistema de degradación de la TOPI, por el contrario, parece ser un sistema de estimulación mediante la relocalización de la proteína del nucleoplasma al nucléolo, evitando su degradación proteosomal84.
La regulación pos-traduccional de la TOPI no ha sido aún estudiada en ningún parásito unicelular de interés médico hasta el momento. La ruta de ubiquitinación y degradación proteosomal ha sido descrita en Giardia lamblia85, T. brucei86, T. cruzi87, Leishmania spp.88, Entamoeba spp.89, P. falciparum90 y Toxoplasma gondii91 pero no así los sistemas de SUMOilación y/o reparación. Sin embargo, la mayoría de los genes que codifican las proteínas involucradas en estos mecanismos aparecen anotados como putativos en sus correspondientes Proyectos Genoma, sugiriendo que estos mecanismos de regulación post-traduccional aparecen ya en los microorganismos eucarióticos más primitivos.
En conclusión: la TOPI es una diana establecida frente a ciertos procesos tumorales cuya efectividad frente a enfermedades tropicales causadas por parásitos unicelulares está en fase de investigación. La estructura dimérica, filogenéticamente única de la enzima de tripanosomátidos y su localización dual -asociada tanto al ADN genómico como al ADN de kinetoplasto- la convierte en una atractiva diana para la creación y desarrollo de nuevos fármacos. A pesar del elevado número de inhibidores de la TOPI ensayados frente a células tumorales y de que algunos de estos compuestos se encuentran en fases de evaluación clínica, se dispone de información muy escasa de su efecto terapéutico frente a tripanosomatidos y otros parásitos. Los buenos resultados obtenidos in vitro frente algunos de estos microorganismos así como el reciente descubrimiento de un mecanismo de muerte celular, semejante a la apoptosis del hospedador, inducida por los inhibidores de clase I, urgen a ensayar infecciones experimentales que justipre-cien la valía de estos compuestos como antibióticos antiprotozoarios.
Agradecimientos: Los autores agradecen a Rebeca Gutiérrez de Prado y a Celia Fernández Rubio por la ayuda prestada en la redacción del presente artículo. Parte de la investigación realizada por los autores y que forma parte de algunos de los artículos incluidos en esta revisión, ha sido financiada por la Comisión Interministerial de Ciencia y Tecnología, CICYT (grants AGL2003 06976/GAN y AGL2003 07420/GAN) del Ministerio de Educación y Ciencia y por el Fondo de Investigaciones Sanitarias (grant PI06302) del Ministerio de Sanidad y Consumo del Reino de España.
1. Greenwood BM, Bojang K, Whitty CJM, Targett GAT. Malaria. Lancet 2005; 365: 1487-98.
2. Shlomai J. The structure and replication of kinetoplast DNA. Curr Mol Med 2004; 4: 623-47.
3. Kioy D, Jannin J, Mattock N. Human African trypanosomiasis. Nat Rev Microbiol 2004; 2: 186-7.
4. Miles MA, Feliciangeli MD, de Arias AR. American trypanosomiasis (Chagas' disease) and the role of molecular epidemiology in guiding control strategies. BMJ 2003; 326: 1444-8.
5. Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis 2004; 27: 305-8
6. May J, Meyer CG. Chemoresistance in falciprum malaria. Trends Parasitol 2003 19; 432-5.
7. Le Bras J, Durand R. The mechanisms of resistance to antimalarial drugs in Plasmodium falciparum. Fundam Clin Pharmacol 2003; 17: 147-53.
8. Ashley EA, White NJ. Artemisinin-based combinations. Curr Opin Infect Dis 2005; 18: 531-6.
9. Legros D, Ollivier G, Gastellu-Etchegorry M, Paquet C, Burri C, Jannin J, et al. Treatment of human African trypanosomiasis-present situation and needs for research and development. Lancet Infect Dis 2002; 2: 437-40.
10. Kennedy PG. Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest 2004; 113: 496-504.
11. Mpia B, Pepin J. Combination of eflornithine and melarsoprol for melarsoprol-resistant Gambian trypanosomiasis. Trop Med Int Health 2002; 7: 775-9.
12. Urbina JA, Docampo R. Specific chemotherapy of Chagas' disease: controversies and advances. Trends Parasitol 2003: 19; 495-501.
13. Docampo R. Sensitivity of parasites to free radical damage by antiparasitic drugs. Chem Biol Interact 1990; 73: 1-27.
14. Croft SL, Coombs GH. Leishmaniasis - current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol 2003; 19: 502-8
15. Balaña-Fouce R, Reguera RM, Cubría JC, Ordóñez D. The pharmacology of leishmaniasis. Gen Pharmacol 1998; 30: 435-43.
16. Bommer W, Eibl HJ, Engel KR, Kuhlencord A, Sindermann H, Sundar S, et al. Leishmaniasis - oral treatment with hexadecylphosphocholine. Wien Klin Wochenschr 2004; 116: 24-9.
17. Rivas L, Moreno J, Cañavate C, Alvar J. Virulence and disease in leishmaniasis: what is relevant for the patient? Trends Parasitol 2004; 20: 297-301.
18. Harms G, Feldmeier H. The impact of HIV infection on tropical diseases. Infect Dis Clin North Am 2005; 19: 121-35.
19. Burton A. Sharing needles may produce artificial Leishmania cycle. Lancet Infect Dis 2001; 1: 4.
20. Vinograd J, Lebowitz J, Radloff R, Watson R, Laipis P. The twisted circular form of polyoma viral DNA. Proc Natl Acad Sci USA 1965; 53: 1104-11.
21. Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 2001; 70: 369-413.
22. Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 2002; 3: 430-40.
23. Corbett KD, Berger JM. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu Rev Biophys Biomol Struct 2004; 33: 95-118.
24. Wang JC. DNA topoisomerases. Annu Rev Biochem 1996; 65: 635-92.
25. Cheesman S, McAleese S, Goman M, Johnson D, Horrocks P, Ridley RG, et al. The gene encoding topoisomerase II from Plasmodium falciparum. Nucleic Acids Res 1994; 22: 2547-51.
26. Christopher LJ, Dykstra CC. Identification of a type II topoisomerase gene from Cryptosporidium parvum. J Eukaryot Microbiol 1994; 41: 28S.
27. Strauss PR, Wang JC. The TOP2 gene of Trypanosoma brucei: a single-copy gene that shares extensive homology with other TOP2 genes encoding eukaryotic DNA topoisomerase II. Mol Biochem Parasitol 1990; 38: 141-50.
28. Fragoso SP, Goldenberg S. Cloning and characterization of the gene encoding Trypanosoma cruzi DNA topoisomerase II. Mol Biochem Parasitol 1992; 55: 127-34.
29. Das A, Dasgupta A, Sharma S, Ghosh M, Sengupta T, Bandopadhyay S, et al. Characterization of the gene encoding type II DNA topoisomerase from Leishmania donovani: a key molecular target in antileishmanial therapy. Nucleic Acids Res 2001; 29: 1844-51.
30. Hanke T, Ramiro MJ, Trigueros S, Roca J, Larraga V. Cloning, functional analysis and post-transcriptional regulation of a type II DNA topoisomerase from Leishmania infantum. A new potential target for anti-parasite drugs. Nucleic Acids Res 2003; 31: 4917-28.
31. Stewart L, Redinbo MR, Qiu X, Hol WG, Champoux JJ. A model for the mechanism of human topoisomerase I. Science 1998; 279: 1534-41.
32. Champoux JJ. Domains of human topoisomerase I and associated functions. Prog Nucleic Acids Res Mol Biol 1998; 60: 111-32.
33. Stewart L, Ireton GC, Champoux JJ. The domain organization of DNA topoisomerase I. J Biol Chem 1996; 271: 7602-8.
34. Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WG. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998; 279: 1504-13.
35. Riou JF, Gabillot M, Philippe M, Schrevel J, Riou G. Purification and characterization of Plasmodium berghei DNA topoisomerases I and II: drug action, inhibition of decatenation and relaxation, and stimulation of DNA cleavage. Biochemistry 1986; 25: 1471-9.
36. Tosh K, Kilbey B. The gene encoding topoisomerase I from the human parasite Plasmodium falciparum. Gene 1995; 163: 151-4.
37. Tosh K, Cheesman S, Horrocks P, Kilbey B. Plasmodium falciparum: stage-related expression of topoisomerase I. Exp Parasitol 1999; 91: 126-32.
38. Villa H, Otero-Marcos AR, Reguera RM, et al. A novel active DNA topoisomerase I in Leishmania donovani. J Biol Chem 2003; 278: 3521-6.
39. Bodley AL, Chakraborty AK, Xie S, Burri C, Shapiro TA. An unusual type IB topoisomerase from African trypanosomes. Proc Natl Acad Sci USA 2003; 100: 7539-44.
40. Das BB, Sen N, Ganguly A, Majumder HK. Reconstitution and functional characterization of the unusual bi-subunit type I DNA topoisomerase from Leishmania donovani. FEBS Lett 2004; 565: 81-8.
41. Wang Z, Englund PT. RNA interference of a trypanosome topoisomerase II causes progressive loss of mitochondrial DNA. EMBO J 2001; 20: 4674-83.
42. Bakshi RP, Shapiro TA. RNA interference of Trypanosoma brucei topoisomerase IB: both subunits are essential. Mol Biochem Parasitol 2004; 136: 249-55.
43. Das A, Dasgupta A, Sengupta T, Majumder HK. Topoisomerases of kinetoplastid parasites as potential chemotherapeutic targets. Trends Parasitol 2004; 20: 381-7.
44. Balaña-Fouce R, Redondo CM, Pérez-Pertejo Y, Díaz- González R, Reguera RM. Targeting atypical trypanosomatid DNA topoisomerase I. Drug Discov Today 2006;11: 733-40.
45. Reguera RM, Redondo CM, Gutiérrez de Prado R, Pérez- Pertejo Y, Balaña-Fouce R. DNA topoisomerase I from parasitic protozoa: a potential target for chemotherapy. Biochim Biophys Acta 2006; 1759: 117-31.
46. Wang JC. DNA topoisomerases as targets of therapeutics: an overview. Adv Pharmacol 1994; 29: 1-19.
47. Capranico G, Giaccone G, Zunino F, Garattini S, D'Incalci M. Development of DNA topoisomerase-related therapeutics: a short perspective of new challenges. Curr Med Chem Anticancer Agents 2004; 4: 335-45.
48. Pizzolato JF, Saltz LB. The camptothecins. Lancet 2003; 361: 2235-42.
49. Adams DJ. The impact of tumor physiology on camptothecin- based drug development. Curr Med Chem Anticancer Agents 2005; 5: 1-13.
50. Sriram D, Yogeeswari P, Thirumurugan R, Bal TR. Camptothecin and its analogues: a review on their chemotherapeutic potential. Nat Prod Res2005; 19: 393- 412.
51. Bodley AL, Shapiro TA. Molecular and cytotoxic effects of camptothecin, a topoisomerase I inhibitor, on trypanosomes and Leishmania. Proc Natl Acad Sci USA 1995; 92: 3726-30.
52. Bodley AL, Cumming JN, Shapiro TA. Effect of camptothecin, a topoisomerase I inhibitor on Plasmodium falciparum. Biochem Pharmacol 1998; 55: 709-11.
53. Bodley AL, Wani MC, Wall ME, Shapiro TA. Antitrypanosomal activity of camptothecin analogs. Structureactivity correlations. Biochem Pharmacol 1995; 50: 937- 42.
54. Staker BL, Feese MD, Cushman M, et al. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem 2005; 48: 2336-45.
55. Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin Jr AB, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA 2002; 99: 15387-92.
56. Redinbo MR, Champoux JJ, Hol WG. Structural insights into the function of type IB topoisomerases. Curr Opin Struct Biol 1999; 9: 29-36.
57. Lee N, Bertholet S, Debrabant A, Muller J, Duncan R, Nakhasi HL. Programmed cell death in the unicellular protozoan parasite Leishmania. Cell Death Differ 2002; 9: 53-64.
58. Sen N, Das BB, Ganguly A, Mukherjee T, et al. Camptothecin induced mito-chondrial dysfunction leading to programmed cell death in unicellular hemoflagellate Leishmania donovani. Cell Death Differ 2004; 11: 924-36.
59. Sen N, Das BB, Ganguly A, Mukherjee T, Bandyopadhyay S, Majumder HK. Camptothecin-induced imbalance in intracellular cation homeostasis regulates programmed cell death in unicellular hemoflagellate Leishmania donovani. J Biol Chem 2004; 279: 52366-75.
60. Proulx M.E, Desormeaux A, Marquis JF, Olivier M, Bergeron MG. Treatment of visceral leishmaniasis with sterically stabilized liposomes containing camptothecin. Antimicrob Agents Chemother 2001; 45: 2623-7.
61. Meng LH, Liao ZY, Pommier Y. Non-camptothecin DNAtopoisomerase I inhibitors in cancer therapy. Curr Top Med Chem 2003; 3: 305-20.
62. Deterding A, Dungey FA, Thompson KA, Steverding D. Anti-trypanosomal activities of DNA topoisomerase inhibitors. Acta Trop 2005; 93: 311-6.
63. Antony S, Kohlhagen G, Agama K, et al. Cellular topoisomerase I inhibition and antiproliferative activity by MJIII- 65 (NSC 706744), an indenoisoquinoline topoisomerase I poison. Mol Phar-macol 2005; 67: 523-30.
64. Hazra B, Saha AK, Ray R, Roy DK, Sur P, Banerjee A. Antiprotozoal activity of diospyrin towards Leishmania donovani promastigotes in vitro. Trans R Soc Trop Med Hyg 1987; 81: 738-41.
65. Ray S, Hazra B, Mittra B, Das A, Majumder HK. Diospyrin, a bisnaphthoquinone: a novel inhibitor of type I DNA topoisomerase of Leishmania donovani. Mol Pharmacol 1998; 54: 994-9.
66. Syrovets T, Buchele B, Gedig E, Slupsky JR, Simmet T. Acetyl-boswellic acids are novel catalytic inhibitors of human topoisomerases I and II. Mol Pharmacol 2000; 58: 71-81.
67. Chowdhury AR, Mandal S, Goswami A, et al. Dihydrobetulinic acid induces apoptosis in Leishmania donovani by targeting DNA topoisomerase I and II: implications in anti-leishmanial therapy. Mol Med 2003; 9: 26-36.
68. Vennerstrom J, Lovelace JK, Waits VB, Hanson WL, Klayman DL. Berberine derivatives as antileishmanial drugs. Antimicrob Agents Chemother 1990; 34: 918-21.
69. Marquis JF, Makhey D, LaVoie EJ, Olivier M. Effects of topoisomerases inhibitors protoberberine on Leishmania donovani growth, macrophage function, and infection. J Parasitol 2003; 89: 1048-52.
70. Lucumi A, Robledo S, Gama V, Saravia NG. Sensitivity of Leishmania viannia panamensis to pentavalent antimony is correlated with the formation of cleavable DNA-protein complexes. Antimicrob Agents Chemother 1998; 42: 1990-5.
71. Chakraborty AK, Majumder HK. Mode of action of pentavalent antimonials: specific inhibition of type I DNA topoisomerase of Leishmania donovani. Biochem Biophys Res Commun 1988; 152: 605-11.
72. Walker J, Saravia NG. Inhibition of Leishmania donovani promastigote DNA topoisomerase I and human monocyte DNA topoisomerases I and II by antimonial drugs and classical antitopoisomerase agents. J Parasitol 2004; 90: 1155-62.
73. Chen AY, Yu C, Gatto B, Liu LF. DNA minor groovebinding ligands: a different class of mammalian DNA topoisomerase I inhibitors. Proc Natl Acad Sci USA 1993; 90: 8131-5
74. Jean-Moreno V, Rojas R, Goyeneche D, Coombs GH, Walker J. Leishmania donovani: differential activities of classical topoisomerase inhibitors and antileishmanials against parasite and host cells at the level of DNA topoisomerase I and in cytotoxicity assays. Exp Parasitol 2006; 112: 21-30.
75. Baguley BC, Ferguson LR. Mutagenic properties of topoisomerase-targeted drugs. Biochim Biophys Acta 1998; 1400: 213-22.
76. Pourquier P, Pommier Y. Topoisomerase I-mediated DNA damage. Adv Cancer Res 2001; 80: 189-216.
77. Pommier Y, Redon C, Rao VA, et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat Res 2003; 532: 173-203.
78. Desai SD, Liu LF, Vazquez-Abad D, D'Arpa P. Ubiquitindependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J Biol Chem 1997; 272: 24159-64.
79. Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, Liu LF. Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res 2001; 61: 5926-32.
80. Pommier Y. Camptothecins and topoisomerase I: a foot in the door. Targeting the genome beyond topoisomerase I with camptothecins and novel anticancer drugs: importance of DNA replication, repair and cell cycle checkpoints. Curr Med Chem Anticancer Agents 2004; 4: 429-34.
81. Cusack Jr. JC, Liu R, Houston M, et al. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS- 341: Implications for systematic nuclear factor-kB inhibition. Cancer Res 2001; 61: 3535-40.
82. Mao Y, Sun M, Desai SD, Liu LF. SUMO-1 conjugation to topoisomerase I: A possible repair response to topoisomerase-mediated DNA damage. Proc Natl Acad Sci USA 2000; 97: 4046-51.
83. Horie K, Tomida A, Sugimoto Y, et al. SUMO-1 conjugation to intact DNA topoisomerase I amplifies cleavable complex formation induced by camptothecin. Oncogene 2002; 21: 7913-22.
84. Mo YY, Yu Y, Shen Z, Beck WT. Nucleolar delocalization of human topoisomerase I in response to topotecan correlates with sumoylation of the protein. J Biol Chem 2002; 277: 2958-64.
85. Emmerlich V, Santarius U, Bakker-Grunwald T, Scholze H. Isolation and subunit composition of the 20S proteasome of Giardia lamblia. Mol Biochem Parasitol 1999; 100: 131-4.
86. Yao Y, Toth CR, Huang L, et al. Alpha5 subunit in Trypanosoma brucei protea-some can self-assemble to form a cylinder of four stacked heptamer rings. Biochem J 1999; 344: 349-58.
87. Diego JL, Katz JM, Marshall P, et al. The ubiquitinproteasome pathway plays an essential role in proteolysis during Trypanosoma cruzi remodeling. Biochemistry 2001; 40: 1053-62.
88. Robertson CD. The Leishmania mexicana proteasome. Mol Biochem Parasitol 1999; 103: 49-60.
89. Hellberg A, Sommer A, Bruchhaus I. Primary sequence of a putative non-ATPase subunit of the 26S proteasome from Entamoeba histolytica is similar to the human and yeast S2 subunit. Parasitol Res 1999; 85: 417-20.
90. Certad G, Abrahem A, Georges E. Cloning and partial characterization of the proteasome S4 ATPase from Plasmodium falciparum. Exp Parasitol 1999; 93: 123-31.
91. Paugam A, Creuzet C, Dupouy-Camet J, Roisin MP. Evidence for the existence of a proteasome in Toxoplasma gondii: intracellular localization and specific peptidase activities. Parasite 2001; 8: 267-73.
Recibido: 12-12-2006
Aceptado: 6-08-2007

