










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.67 n.6-1 supl.1 Ciudad Autónoma de Buenos Aires 2007
Síndrome de Rett: 50 años de historia de un trastorno aun no bien conocido
Jaime Campos-Castello, Daniel M. Fernandez-Mayoralas, Nuria Muñoz-Jareño, Victoria San Antonio-Arce
Servicio de Neurología Pediátrica, Hospital Clínico Universitario San Carlos, Madrid, España
Dirección postal: Dr. Jaime Campos-Castelló, Servicio de Neurología Pediátrica. Hospital Clínico Universitario San Carlos, Profesor Martín Lagos S/N. 28040 Madrid, España Fax: +34 91 330 3553 e-mail: jcampos.hcsc@salud.madrid.org
Resumen
Desde que fue descrito por primera vez por Andreas Rett hace 50 años, el síndrome de Rett (SR) ha sido objeto de muchas investigaciones, sin embargo continúa siendo un trastorno aún no bien conocido. Presentamos nuestra propia experiencia y una revisión de la literatura sobre el SR. Se trata de un trastorno del neurodesarrollo, dominante ligado a X, que afecta casi siempre a mujeres, la mayoría de los casos de forma esporádica. El diagnóstico de SR debe hacerse en base a la observación clínica. Las principales características son la aparición de un retraso mental, cambios conductuales, estereotipias, pérdida del lenguaje y, sobre todo, del uso propositivo de las manos, aparición de una apraxia de la marcha, presencia de alteraciones de la respiración y, frecuentemente, crisis epilépticas. Los criterios diagnósticos consensuados internacionalmente son aquí revisados. El SR se debe en la mayoría de casos a mutaciones del gen MECP2, si bien una proporción de casos atípicos puede estar causada por mutaciones de CDKL5, particularmente la variante con epilepsia precoz. Sin embargo, los mecanismos patogénicos moleculares no son bien conocidos, así como la relación entre las mutaciones de MECP2 y otros trastornos del desarrollo. Revisamos también los hallazgos de neuroimagen, neuropatológicos y neurobioquímicos descritos en el SR. Respecto al tratamiento, aparte del sintomático, no hay ninguno que se haya mostrado eficaz. Un trabajo reciente abre perspectivas terapéuticas futuras al haber demostrado mediante un modelo animal de ratón la reversión de los síntomas neurológicos mediante la activación de la expresión de MeCP2.
Palabras clave: Síndrome de Rett; Criterios diagnósticos; Genética; Diagnóstico molecular; Patogénesis; Neuropatología
Abstract
Rett syndrome: 50 years' history of a still not well known condition. Since it was first described by Andrea Rett 50 years ago, Rett syndrome (RS) has been the subject of further investigations, nonetheless it continues to be a not well known condition. Our own experience and an updated literature review on RS is presented. RS is a severe dominant X chromosome-linked neurodevelopmental disorder with a characteristic clinical picture that mostly occurs in girls, most of the cases are sporadic and genetically determined. The diagnosis of RS is made based on observation and clinical assessment. Main clinical features are mental retardation, behavioural changes, stereotypes, loss of speech and hand skills, gait apraxia, irregular breathing with hyperventilation while awake, and frequent seizures. The internationally established criteria are reviewed. RS is caused by mutations in MECP2 in the majority of cases, but a proportion of atypical cases may result from mutations in CDKL5, particularly the early onset seizure variant. However, the molecular pathogenesis of this disorder remains unclear, as well as the relation between the mutations in MECP2 and other neurodevelopmental disorders. Neuroimaging, neuropathological and biochemical findings in RS are reviewed. Besides symptomatic treatment, no therapeutic trials have shown effectiveness. Some perspectives in the treatment of RS have been provided by a recent work showing a phenotypic reversal by activation of MeCP2 expression in a mouse model.
Key words: Rett syndrome; Diagnostic criteria; Genetics; Molecular diagnosis; Pathogenesis; Neuropathology
El Síndrome de Rett (SR) fue descrito en 1966 por el austríaco Andreas Rett como una atrofia cerebral con hiperamoniemia ("Über ein eigenartiges hirnatrophisches Syndrom bei Hyperammonämie im Kindesalter") propio del sexo femenino. Más tarde, en 1982, se presentó su descripción clínica detallada en la reunión de la Federación Europea de Neuropediatría celebrada en Norwirherhout (Holanda) y, en 1983, Hagberg, Aicardi, Días y Ramos publicaron 35 casos de procedencia sueca, francesa y portuguesa, constituyéndose a partir de entonces como un proceso mejor conocido entre la comunidad científica1, 2. Hoy en día sabemos que el SR posee un marcador biológico en la mutación del gen MECP2, que afecta fundamentalmente a niñas de todas las razas y que es una de las causas conocidas más frecuentes de retraso mental grave en el sexo femenino3 y de manera excepcional en el masculino.
La prevalencia no es bien conocida, pero probablemente oscile entre 0.5 y 1 de cada 10.000 habitantes, es decir, posiblemente superior a la de la fenilcetonuria3. En España, que contaba en 1999 con 39.580.600 habitantes, se pudo localizar 207 casos afectados de SR en un estudio en la población española publicado en dicho año, lo que supone un registro muy bajo de casos4. En nuestro servicio hemos observado hasta la actualidad (2006) 52 casos correspondientes a 51 niñas y un varón, siendo objeto 15 de ellos de la primera publicación española en 19885.
Clínica
El diagnóstico es clínico, dado que sólo un 70 a 80% del fenotipo clínico de SR tiene mutaciones en el gen MECP2 y viceversa, no todas las pacientes con mutaciones en el gen MECP2 tienen SR. Los criterios diagnósticos fueron señalados en 1988 (Tabla 1)6, pero fueron revisados en la reunión llevada a cabo por la European Paediatric Neurology Society en Baden Baden, Alemania, el 11 de septiembre de 2001 (Tabla 2)7. De forma similar, en dicha reunión se redactaron los criterios de las variantes fenotípicas para permitir una mayor inclusión de pacientes (Tabla 3).
TABLA 1.- Criterios de diagnóstico para el SR clásico6
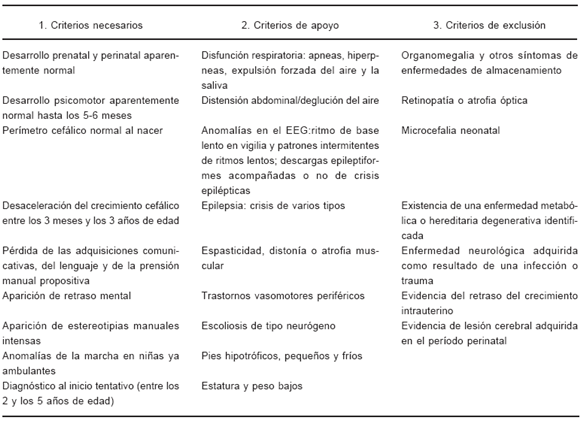
TABLA 2.- Criterios de diagnóstico para el SR clásico -Revisados (Baden Baden)7
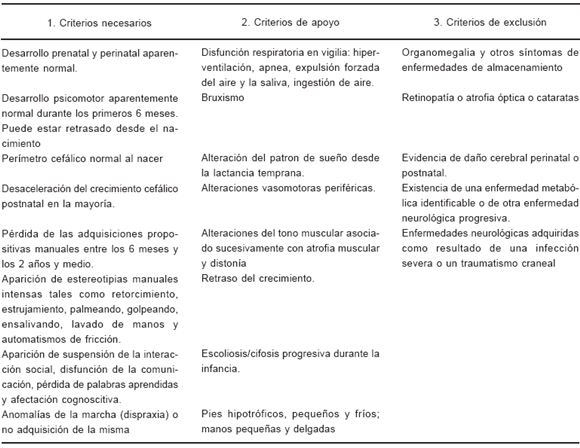
TABLA 3.- Delineación revisada de las variantes fenotípicas (Baden Baden)7

El cuadro clínico cursa con un período prenatal y perinatal normal, aunque hay formas "congénitas", y un desarrollo normal o casi normal durante los primeros meses de vida. Tras este período, entre los 3 meses y 3 años de vida se pierden las capacidades manuales propositivas, uno de los elementos más característicos de la enfermedad, y se produce una regresión de las funciones psicomotoras y de la comunicación. El contacto ocular está muy limitado e incluso puede ser, curiosamente, intenso y "vivo" (al revés que en la mayoría de pacientes autistas), lo que a veces permite cierta capacidad de comunicación con la familia3, 5. Aunque es normal al nacimiento, el crecimiento del perímetro craneal sufre un descenso paulatino (que puede conducir a microcefalia), que puede ser tan precoz como en los tres primeros meses de vida, constituyendo a veces el primer signo del SR. Entre el primer y tercer año de edad aparece la seña de identidad del SR: los movimientos estereotipados de las manos, característicamente de lavado (manos juntas), pero también de palmoteo o de aplausos (manos separadas), sobre todo en la línea media1, 3, 5. Pueden aparecer otras estereotipias y "pseudocrisis" (temblores, caídas bruscas, detención del movimiento, episodios de risa o gritos inmotivados)5. La marcha suele ser normal al inicio pero se va volviendo apráxica, amplia, errática y no propositiva. Con frecuencia la marcha se inicia dando pasos hacia atrás (retropulsión). Es frecuente el balanceo de un lado a otro. Algunos pacientes presentan, desde el inicio de la enfermedad, alteraciones neurológicas evidentes, tales como ataxia del tronco, escoliosis neurógena, crisis convulsivas, alteraciones de la respiración, alteraciones gastrointestinales, disfunción piramidal, hipoacusia neurosensorial leve3, neuropatía periférica (detectable por EMG en aproximadamente la mitad de los pacientes, y con características de neuropatía axonal)8 o distonía, pero habitualmente dichos signos se presentan posteriormente, de forma progresiva, predominando en los primeros estadios de la enfermedad un trastorno generalizado del desarrollo capaz de simular un autismo. De hecho, debido al característico fenotipo conductual, en el DSM-IV el SR forma parte de los trastornos de inicio en la infancia, la niñez o la adolescencia bajo el epígrafe de "Trastornos generalizados del desarrollo" - "Trastorno de Rett" (299.80). A nuestro juicio, los criterios de inclusión son netamente inferiores a los expuestos previamente y, además, el SR debe ser considerado como un trastorno neurológico y no mental o psiquiátrico5.
Las pacientes con SR tienen mayor riesgo de muerte s úbita. Una razón puede ser el riesgo de arritmias cardíacas, incluida la prolongación del segmento QT y la bradicardia severa que puede requerir el implante de un marcapasos3. Aunque se ha descrito la asociación de SR y diabetes mellitus aún no está clara la naturaleza de dicha asociación, que podría ser casual9.
La invalidez es importante, y la falta de cuidados lleva caracter ísticamente a estas pacientes a la escoliosis deformante y a la desnutrición grave, por lo que la expectativa de vida está disminuida a pesar de que existen raros casos que han sobrepasado los 60 años, y a pesar de que se ha producido un incremento en la longevidad de estas pacientes durante los últimos años (Fig. 1).

Fig. 1.- Síndromes de Rett de más de 21 años en 2002 (Fuente: IRSA).
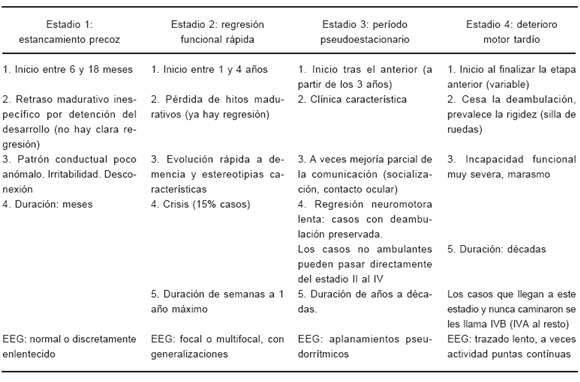
De forma didáctica se han establecido cuatro estadios clínicos10, 11. Los cambios de una etapa a otra se producen a lo largo de un "continuum" más que de forma abrupta (Tabla 4). De hecho, la progresión del SR, que inicialmente se catalogó como progresiva salvo algunos casos que podían mejorar llegada la edad adulta, muestra una rápida sucesión de síntomas en los dos primeros estadios, cierto estancamiento e incluso alguna mejoría en aspectos de la socialización al llegar al estadio III, progresando o evolucionando los aspectos motores, la escoliosis, la apraxia y ataxia de la marcha y las estereotipias manuales, haciéndose más evidentes los movimientos distónicos, las alteraciones de la respiración, la epilepsia (rebelde al tratamiento), el déficit de crecimiento, las alteraciones gastrointestinales y la disfunción autonómica. Al llegar a la edad adulta, la mujer con SR habitualmente se encuentra en una situación de invalidez motora, bien bajo una forma rígido-distónica o bajo forma hipotónico-atrófica; pero el deterioro mental previamente alcanzado (así como el decremento del perímetro craneal) no progresa y queda estacionario, y en esta evolución mejoran la epilepsia, las anomalías respiratorias y la socialización.
TABLA 4.- Estadios clínicos del SR3, 5, 8, 10
Epilepsia, EEG y SR
Como ocurre en otras epilepsias sintomáticas, es frecuente que coincidan varios tipos de crisis en la misma paciente, y los tipos de crisis son muy variados; las más frecuentes son las crisis tónicas seguidas por las ausencias (que pueden ser atípicas), crisis atónicas, crisis parciales simples o complejas, generalizadas tónico-clónicas y fotosensibles entre otras5, 8, 12. Los hallazgos EEG principales consisten en actividad de fondo lenta y en actividad paroxística (suele desarrollarse en estadio II, sobre todo a partir de los tres años) durante el sueño, multifocal en las más jóvenes y focal en las más mayores. Estas actividades, persistentes y prácticamente constantes en las pacientes después de los dos años de edad, aparecen independientemente de la existencia de crisis clínicas, exacerbándose en fase de sueño N-REM, donde pueden adquirir un aspecto de generalización de punta onda-lenta (2 Hz). También se ha observado una disminución del sueño REM (situación observada en pacientes con disfunción cognoscitiva). Es menos frecuente (25% casos) el hallazgo de una actividad lenta monomorfa de gran amplitud (>150 mV) interrumpida por fases de aplanamiento pseudoperiódico8. La actividad paroxística, así como las crisis clínicas, van disminuyendo en el estadio III y IV, aunque pueden persistir trazados paroxísticos o el trazado de base evoluciona a un enlentecimiento progresivo del ritmo de base según avanzan los estadios clínicos. Los trastornos electroencefalográficos del SR según los estadios clínicos aparecen resumidos en la Tabla 4. En ocasiones el EEG es semejante al típico del Síndrome de Angelman (SA), caracterizado por puntaonda lenta de gran amplitud (normalmente 2-3 Hz), sobre todo en regiones posteriores y facilitado por el cierre palpebral3, 13. Puede ser difícil diferenciar patrones conductuales de crisis comiciales sin monitorización vídeo-EEG.
Variantes del SR
Las niñas que cumplen con la mayoría de los criterios de diagnóstico del SR son clasificadas como pacientes con SR clásico. No obstante, el fenotipo es más variado de lo que se describió al principio. La mayoría de estas variantes son, comparadas con la forma clásica, más leves, especialmente en el grado de disfunción motora. Hagberg y colaboradores describieron cinco variantes11, que se asocian con mucha menor frecuencia que la forma clásica a mutaciones del gen MECP2, y que recientemente se están viendo asociadas a otros genes relacionados con el anterior 45.
1. Forma con epilepsia precoz: son pacientes que cumplen con los criterios del SR clásico pero inician los síntomas antes de los 6 meses de edad y la presentación clínica inicial está dominada por las crisis epilépticas (que suelen manifestarse bajo la semiología de espasmos infantiles con hipsarritmia) lo que enmascara el diagnóstico5. Esta forma clínica parece estar producida por mutaciones en el gen CDKL5 que codifica una proteína- quinasa nuclear cuya expresión en el sistema nervioso se superpone a la de MeCP2 (con la que interactúa mediante fosforilación), durante la maduración neuronal y la sinaptogénesis14.
2. Forma congénita: No existe un período de normalidad en el desarrollo psicomotor, pero se cumplen los criterios del SR clásico. Cada vez se describen más casos.
3. Forma de regresión tardía: la regresión del desarrollo psicomotor se manifiesta tardíamente (generalmente después de los 4 años de edad) y de forma más insidiosa.
4. Forma "frustra": curso clínico leve e incompleto. La regresión se produce habitualmente entre el primer y el tercer año de vida, pero es más leve que en el SR clásico. El uso de las manos puede estar preservado y las estereotipias pueden ser mínimas o atípicas. No suelen presentar microcefalia ni talla baja.
5. Forma con conservación del lenguaje: tras la fase de regresión, consiguen pronunciar algunas palabras que fueron aprendidas antes de manifestarse la enfermedad (nunca adquiridas después de la fase de regresión). Algunas de estas pacientes tienen mutaciones en el gen MECP2, al igual que la mayoría de las pacientes con la forma clásica del SR.
No obstante, existen otros muchos fenotipos dependientes de las diferentes mutaciones de MECP2 así como de la inactivación sesgada o no del cromosoma X (Tabla 5).
TABLA 5.- Fenotipos relacionados con mutaciones de MECP23

SR en el sexo masculino
Los casos descritos corresponden a niños con cariotipo 47 XYY15 y a hermanos de niñas con SR familiar, que provoca frecuentemente encefalopatías letales en la infancia. Excepcionalmente, el SR en niños puede manifestarse como una encefalopatía no letal con un curso semejante al del sexo femenino16 (Tabla 5). La clínica en estos casos se ha señalado que puede diferir de la clásica que muestran las niñas, pero en nuestro caso personal no existió esta diferencia.
Diagnóstico diferencial (Tabla 6)
TABLA 6.- Diagnóstico diferencial del SR17,46

Aunque el diagnóstico habitualmente no es difícil queremos insistir en las dificultades que en ocasiones plantea el Síndrome de Angelman (SA) en pacientes del sexo femenino, así como la inestimable ayuda que nos proporcionan los tests genéticos para la correcta ubicación nosológica del trastorno 17. El diagnóstico diferencial entre el autismo de Kanner y el SR se muestra en la Tabla 7.
TABLA 7.- Diagnóstico diferencial de la conducta autista5

Bases genéticas y fisiopatológicas
El SR es una enfermedad dominante ligada al sexo, casi siempre letal en los varones, producida por la mutación heterocigota del gen MECP2, situado en la región distal del cromosoma X (Xq28), que codifica una proteína llamada MeCP2 (proteína 2 de fijación de la metil-citosinaguanosina o methyl-CpG-binding repressor protein 2) la cual se une al DNA y tiene la función de silenciar otros genes3, 18, 19. Existen varias isoformas de esta proteína20. MeCP2 se expresa en todo el organismo, pero con más intensidad en el cerebro. Aproximadamente un 85% de las afectadas de SR clásico (y un porcentaje inferior al 50% de formas atípicas) presenta mutaciones en la región codificante del gen MECP23. La mayor parte de las veces, las mutaciones son "de novo", por lo que el SR se presenta de forma esporádica en el 99% de los casos, y el riesgo de la pareja de tener otra hija afectada es inferior al 1%3. La correlación entre el fenotipo y el genotipo se creía que dependía fundamentalmente de la variabilidad interindividual en la inactivación aleatoria del cromosoma X, así como de la posición y tipo de mutación3, 21. Cada vez se está dando menos importancia a la dicha inactivación y más importancia a los tipos de mutación, como así lo demuestran los resultados de los trabajos más recientes22, 23. En la Fig. 2 pueden verse algunas de las mutaciones del gen en nuestra casuística.

Fig. 2.- Algunas de las mutaciones del gen MECP2 en nuestra casuística.
Se ha identificado más de 225 mutaciones en el gen MECP2, aunque en el 70% de los SR que cumplen completamente los criterios de consenso se encuentran tan sólo ocho. La gran mayoría de las mutaciones representan cambios de un único nucleótido, pero entre las mutaciones específicas las deleciones o inserciones a pequeña escala representan el 60% de las mismas, las primeras agrupadas en la región C-terminal de MECP23. Las pacientes con mutaciones de tipo "missense" padecen habitualmente un síndrome menos severo con mayor perímetro craneal y mejor desarrollo del lenguaje que aquellas con mutaciones de tipo "nonsense". La región del gen afectada parece ser menos importante24, 25, no obstante, también puede traducirse en características peculiares del fenotipo. En deleciones del C-terminal la distonía es precoz y sin embargo el comportamiento adaptativo está mejor preservado26. Una mujer con mutación en la región C-terminal tenía retraso mental y epilepsia pero menor afectación manual y del perímetro cefálico27. Una mutación leve que no produce síntomas en mujeres (mutación "missense" en la WW domain binding region) produce retraso mental en varones, por lo que las mutaciones leves en los varones son suficientes para producir alteraciones importantes20, 21
La mayor ía de uniones de MeCP2 se realizan con secuencias DNA silenciadas a través de la metilación28. La regulación exquisita de MeCP2 es de vital importancia, puesto que a través de animales de laboratorio se ha observado patología grave tanto por exceso como por defecto en la producción de dicha proteína y a edades diferentes20, 30. Por ejemplo un ratón de laboratorio transgénico capaz de expresar la proteína MeCP2 en exceso parecía tener mayor capacidad de aprendizaje las primeras 10 semanas de vida, pero se volvía hipoactivo y desarrollaba crisis convulsivas después31. En otro estudio se comprobó que la sobreexpresión de MeCP2 en ratones producía un aumento de la complejidad dendrítica y de la longitud de los axones en las neuronas corticales, de lo que se ha deducido que MeCP2 regula directamente la maduración neuronal y la sinaptogénesis e influye en las elongaciones dendríticas y los procesos de ramificación a través de diferentes mecanismos32.
La metodolog ía para detectar la mutación se ha estado basando en la amplificación mediante reacción en cadena de polimerasa (PCR) y la subsiguiente secuenciación de los exones codificantes de MECP2. No obstante, este método no detecta deleciones a gran escala o reorganizaciones en MECP2, responsables de al menos el 10% de los falsos negativos en niñas con SR clásico, mediante análisis mediante PCR estándar, por lo que están en desarrollo mejores métodos diagnósticos (análisis mediante Southern blot, PCR cuantitativa o amplificación mediante sonda dependiente de ligamiento múltiple)3. En la Tabla 8 aparecen las indicaciones formales para la realización del test genético de MECP23.
TABLA 8.- Indicaciones para la realización del test genético de MECP23

En los pocos casos familiares de SR se ha descubierto una gran variabilidad en cuanto a las anomal ías fenotípicas dentro de una misma familia, que oscilan entre la ausencia de síntomas, pasando por un trastorno leve del aprendizaje y un SR clásico hasta una encefalopatía letal en un varón3. Por lo tanto, existen muchos fenotipos distintos relacionados con mutaciones de MECP2, y estos fenotipos son posibles en ambos sexos (Tabla 4). Dos varones XY no relacionados con una mutación idéntica en MECP2 (al igual que sus madres asintomáticas) padecían un síndrome caracterizado por deterioro cognoscitivo grave, macrocefalia, diarrea crónica y espasticidad progresiva. Este fenotipo se explica por un mosaicismo somático para MECP23. También se han detectado mutaciones en varones con retraso mental inespecífico cuyo único rasgo común era un retraso en el lenguaje33. En nuestra casuística el caso varón correspondía a un SR típico en el que se confirmó la mutación MECP2.
Función de MECP2
La proteína MeCP2 funciona en el silenciamiento de la transcripción uniéndose a dinucleótidos CpG metilados en regiones promotoras de genes. En el genoma, la mayoría de los residuos de citosina en los dinucleótidos CpG están metilados. Esta silenciación de los genes mediante la metilación es operativa en numerosos procesos celulares, como la inactivación aleatoria de un cromosoma X en el sexo femenino, la regulación de la transcripción y otros procesos. Parece ser que el silenciamiento de la transcripción en las neuronas podría favorecer una función celular eficiente evitando el "exceso de ruido" de la transcripción. En este proceso la MeCP2 interaccionaría con una proteína co-represora (Sin3A) y con la desacetilasa de histonas (HDAC) regulando la transcripción mediante la compactación de la cromatina. En este modelo la MeCP2 se uniría a la metilcitosina, lo que movilizaría la proteína correpresora Sin3A y la HDAC. Esta última, a continuación, desacetilaría los grupos de acetato de la histona, provocando la compactación de la cromatina, lo que a su vez regularía a la baja o inhibiría la transcripción de genes responsables del SR, algunos de los cuales, como veremos a continuación, comienzan a conocerse3. MeCP2 también interaccionaría con la metiltransferasa de DNA y de histona34.
Aunque no se conoce la influencia de la apoptosis en el SR es posible que exista una susceptibilidad disminuida (resistencia) a la misma31.
Síndrome de Rett y otros trastornos del desarrollo
Los signos y síntomas del SR son dependientes de la edad del paciente, y se piensa que la mutación de MECP2 produce una sinaptogénesis anormal en diferentes etapas del desarrollo19, es decir, que se produciría una alteración de la formación, maduración neuronal y del desarrollo de las sinapsis debido a la imposibilidad por parte de un MECP2 mutado de poder suprimir la expresión de genes que deberían ser silenciados durante este proceso33, 36. Aunque la presencia de MeCP2 no requiere la formación de sinapsis, la cantidad de dicha proteína aumenta conforme aumenta la formación de sinapsis. Las neuronas maduras que han formado sinapsis inducen niveles plenos de MeCP2, y en animales de experimentación se ha comprobado que MeCP2 posee una función biológica crítica en las etapas finales del desarrollo neuronal.
El momento de aparición de la proteína MeCP2 se correlaciona con la ontogenia del sistema nervioso central, y la cantidad de la misma va aumentando con la maduración. Durante la infancia se observa un incremento de las neuronas corticales positivas para MeCP2, mientras que las estructuras más profundas (como la formación reticular) muestran un porcentaje constante de neuronas positivas para MeCP2 desde las 35 semanas de edad gestacional20. De esta forma, las regiones neuronales generadas de forma temprana y por lo tanto más maduras expresan más intensamente MeCP2 durante el período postnatal inmediato, y los cambios más significativos en la expresión de MeCP2 se producen en regiones que maduran después (como la corteza, el cerebelo, o el hipocampo), correlacionándose dicho aumento en la expresión con la sinaptogénesis de estas regiones37.
En el SA, que guarda ciertas similitudes clínicas e incluso, a veces, electroencefalográficas13 con el SR, el gen responsable del fenotipo es el UBE3A, que se encuentra en la región 15q11-q13 y que codifica la E6AP-3A (ubiquitin proteína ligasa 3A), importante para la degradación de proteínas en las neuronas34, 38, 39. La deficiencia de MeCP2 parece afectar a UBE3A, y parte del fenotipo del SR se debe al silenciamiento secundario de UBE3A mediante una reducción en la expresión de la copia materna de dicho gen y de su producto proteico40, 41. Es posible que otras proteínas diferentes a UBE3A (BDNF o xhairy 2a) estén afectadas por la deficiencia de MeCP2 e involucradas en los síntomas del SR, aunque este mecanismo explicaría las similitudes clínicas entre el SA y el SR41. Un gen del tipo "nonimprinted" de la región 15q11- q13, llamado GABAR3, que codifica la subunidad beta3 del receptor GABA (gen candidato para el autismo) también se ha visto reducido en su expresión en los mismos síndromes20, 40.
Diversos estudios20, 34, 40, 41 sugieren una relación entre genes relacionados con el SA, el autismo y el gen MECP2. Existe por lo tanto cierta superposición en las diferentes vías que llevan a la mala regulación de los genes de la región 15q11-q13 en el SR, SA y autismo, lo que implica directamente a la proteína MeCP2 en la regulación de la expresión de UBE3A y también de GABAR3 del cerebro humano20, 40. MECP2 no representa, sin embargo, una causa significativa de retraso mental inespecífico42. En algunas ocasiones, mutaciones en MECP2 pueden producir un fenotipo muy parecido al SA (quizás el 2%)34; no obstante, y pese a que está indicado el análisis de MECP2 en los pacientes con fenotipo del SA sin anomalías en 15q11-q13, no parece atribuible a dicha mutación el exceso de pacientes con SA esporádico43, 44.
Existen otros genes candidatos para el SR cuando no está producido por mutaciones en MECP2, uno de ellos es el NTNG1 (Netrina G1), presente en el cromosoma 145.
Neuropatología
El cerebro de la paciente con SR es pequeño. No obstante, y al contrario que en las enfermedades degenerativas del sistema nervioso central, el cerebro no se va haciendo progresivamente más pequeño a lo largo del tiempo de forma indefinida.
Desde hace muchos años se conoce la existencia de un descenso cuantitativo de la melanina en la pars compacta de la sustancia negra33.
Microscópicamente, al igual que en otros muchos trastornos del desarrollo (como el autismo), es posible observar un descenso del tamaño de las neuronas, un incremento en su empaquetamiento y una reducción de las arborizaciones dendríticas. Este descenso en el número de dendritas y sinapsis en áreas selectivas del cerebro (especialmente en la corteza frontal y núcleo caudado, es decir, los mismos lugares donde existe un descenso del volumen) puede deberse a una alteración de la sinaptogénesis o a un "pruning" (eliminación de sinapsis innecesarias) excesivo20, 21, 33. La mutación de MECP2 produce una función postmitótica neuronal incorrecta, que altera la estructura subcortical cerebral, lo que induce anomalías en el comportamiento social instintivo (comportamientos autistas en el SR)21. Como hemos advertido, los hallazgos neuropatológicos descritos son muy similares a los observados en pacientes con Síndrome de Angelman (SA), autismo, Síndrome del cromosoma X frágil e incluso el Síndrome de Down, y algo parecido parece ocurrir con la expresión cerebral reducida del Cterminal de MeCP2, por lo que parece claro que la expresión de MeCP2 está controlada por varias vías diferentes3, 46.
Se piensa que existe una alteración precoz de los sistemas neuronales monoaminérgicos troncoencefálicos, que podría causar anomalías o errores en la sinaptogénesis en el cerebro y también inducción del autismo o rasgos autistas en pacientes con SR (escasa actividad de las neuronas noradrenérgicas y serotonininérgicas con función mejor conservada de las neuronas dopaminérgicas y colinérgicas de la protuberancia). Se piensa que los movimientos manuales estereotipados que se desarrollan después de la pérdida del uso propositivo de las manos (movimiento propiciado por procesos neuroquímicos de refuerzo o consolidación) y la aparición de hipertonía y distonía se deben a la disfunción de los ganglios de la base, la corteza promotora y el área motora suplementaria, con un aumento de la sensibilidad de los receptores dopaminérgicos a través de la vía de proyección tálamocortical (mediada por los ganglios basales). La disfunción dopaminérgica sería secundaria a la afectación de las neuronas monoaminérgicas en el tronco del encéfalo (núcleos pedúnculo-pontinos). Por otro lado, la alteración típica del patrón respiratorio de estas pacientes se corresponde con un centro cardiorrespiratorio anormal en el tronco encefálico y con la afectación del sistema serotoninérgico (20). Probablemente también esté involucrado un descenso de la sustancia P y otros neuromoduladores del tronco del encéfalo. En una serie propia de 20 pacientes con SR, encontramos concentraciones de ácido 5-hidroxindolacético (5-HIAA) en LCR significativamente más bajas que en sujetos control, lo que apoyaría la hipótesis de la existencia de una alteración del metabolismo de la serotonina en el SR47.
Existe una región de genes "imprinted" (una área rica en genes que son transcritos sólo desde el alelo materno o paterno) que contienen el gen Dlx5, cuya transcripción está aumentada en ratones sin la proteína MeCP2 y en células de pacientes con SR y que está relacionada con la producción de enzimas sintetizadores de GABA, por lo que su sobreexpresión puede alterar la actividad neuronal gabaérgica y también formar un circuito o loop silenciador de cromatina nuevo que contribuya al SR48. Quizás exista una puerta abierta a una futura regulación del GABA con algún nuevo producto farmacéutico que mejore los síntomas en estas pacientes.
Tratamiento del SR
No existe un tratamiento específico en la actualidad, por lo que el objetivo es mejorar en lo posible la calidad de vida de la paciente.
La epilepsia puede ser rebelde. No existen antiepilépticos específicamente recomendables en el SR. No obstante, en nuestra experiencia, la carbamazepina en monoterapia o en combinación con clobazam ha sido el tratamiento más eficaz, siendo el ácido valproico un fármaco alternativo (debiendo recordar que puede condicionar un aumento del temblor e hiperamoniemia). En casos rebeldes la vigabatrina ha resultado efectiva5. Respecto a los fármacos más recientes, el topiramato, que tiene efectos GABAérgicos y glutaminérgicos (ambos sistemas parecen involucrados en el SR) ha mostrado cierta eficacia en estudios pequeños, mejorando incluso el patrón respiratorio de algunas pacientes49. La lamotrigina muestra resultados contradictorios, habiendo sido eficaz en algunos casos3. En situaciones de salvas prolongadas de crisis y status la utilización de benzodiazepinas por vía oral o rectal puede resultar de utilidad. En algunos casos de la "forma con epilepsia precoz" hemos utilizado hidrocortisona o ACTH con resultados satisfactorios12. En algún caso la dieta cetogénica ha resultado beneficiosa50. La estimulación vagal para el control de la epilepsia farmacorresistente en el SR se ha mostrado eficaz en alguna serie corta, con 50% de reducción del número de crisis, pero sin modificar la conducta y evolución propia del síndrome.
El riesgo ortopédico está condicionado por los trastornos tónicos de diversa índole que concurren en el SR y que son responsables de la pérdida de la marcha. El inicio precoz de la fisioterapia (fundamental también para prevenir la osteopenia), la hidroterapia al potenciar los movimientos sin el efecto de la gravedad y la hipoterapia para favorecer la postura erecta son métodos que han sido utilizados con eficacia. La escoliosis, problema ortopédico principal, ya que está presente en más del 80% de las pacientes a la edad de 25 años51, se debe anunciar, detectar y combatir desde que se establece el diagnóstico de SR, debido a que es neurogénica y de evolución rápida; aquí de nuevo es eficaz la rehabilitación, la utilización precoz de corsés (aunque diversos trabajos señalan una eficacia limitada) y nosotros somos partidarios de la cirugía relativamente precoz (para algunos autores al alcanzarse una curvatura >40° Cobb), con fusión vertebral temprana por vía posterior, ya que es un método que mejora la calidad de vida en el SR en un alto porcentaje de casos (más del 80%)3.
Para mejorar el estado nutricional, en algunos casos es necesario complementar la dieta tanto en proporciones calóricas como en suplementos vitamínicos; el uso de l-carnitina no se ha mostrado eficaz en nuestra experiencia. Es importante supervisar y tratar el estreñimiento y la salud dental. La incidencia de patología litiásica en vesícula biliar y de problemas gastrointestinales es significativamente más alta que en la población general, por lo que el cambio en el comportamiento o la irritabilidad de la paciente debe llevarnos a realizar un estudio del aparato gastrointestinal y genitourinario3.
El intento de mejorar la clínica mediante la utilización de modificadores de la neurotransmisión ha sido ineficaz en los escasos estudios realizados. Así, los tratamientos con l-dopa/carbidopa o agonistas dopaminérgicos como la bromocriptina y la pergolida han sido ineficaces. Tampoco la utilización de la tirosina y triptófano ha modificado la clínica pese a la mejoría de los niveles de 5-HIIA. La naltrexona (antagonista de las beta-endorfinas) también ha fracasado y aun empeorado el estado general por efecto secundario de sedación41. Ni la busperidona ni el citrato de magnesio han mostrado eficacia en el control de los episodios de hiperventilación y/o apnea. El babeo se puede tratar con parches de escopolamina o con trihexifenidilo (Artane®).
Recientemente se ha reportado un síndrome neurometabólico nuevo caracterizado por un déficit cerebral de folato (CFD) y su tratamiento con ácido folínico, sugiriendo la posibilidad de intentar este tratamiento en el SR, ya que es una causa de CFD secundario52. No se ha demostrado aún la eficacia de dicho tratamiento.
Debe realizarse rehabilitación funcional tanto en lo que se refiere a aspectos fisioterápicos (programas de ejercicio físico con rutinas para personas con SR) como a otras terapias ocupacionales así como metodología de comunicación alternativa. Particularmente el primer aspecto parece obtener excelentes resultados funcionales50. La musicoterapia se ha valorado como eficaz en algunos casos53, 54. Para el control de las estereotipias manuales se ha señalado la utilidad de las férulas de inmovilización55.
Recientemente, y pensando en perspectivas terapéuticas futuras, se ha demostrado mediante un modelo animal de ratón con SR por la mutación de MECP2 que las neuronas sufren cambios durante su maduración a causa del defecto de expresión de MeCP2, pero que no mueren, y que estos cambios pueden ser reversibles mediante la activación de la expresión de MeCP2, con reversión de los síntomas neurológicos tanto en animales inmaduros como en maduros56.
Con vistas a futuros ensayos terapéuticos en el síndrome de Rett, en el último Workshop Internacional realizado en San Francisco, California, en mayo de 2006, se estableció la necesidad de crear un consorcio de investigadores sobre el síndrome de Rett, clínicamente orientados (RettSearch) que recojan la información relevante existente y que asesore los ensayos clínicos, bien individuales bien multicéntricos, propuestos para el síndrome de Rett, bajo el control de la IRSA (Internacional Rett Síndrome Association, http://www.rettsyndrome.org).
Conflicto de interés: ninguno
Bibliografía
1. Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol 1983; 14: 471-9.
2. Hagberg B. Rett syndrome: clinical particularities and biological mysteries. Acta Paediatr Scand 1995; 84: 971-6.
3. Percy AK, Lane JB. Rett syndrome: clinical and molecular update. Curr Opin Pediatr 2004; 16: 670-7.
4. Pineda M, Aracil A, Vernet A, et al. Estudio del síndrome de Rett en la población española. Rev Neurol 1999 Jan 1-15; 28:105-9.
5. Campos-Castelló J, Peral Guerra M, Riviere Gómez A, et al. Síndrome de Rett: estudio de 15 casos. An Esp Pediatr 1988; 4: 286-92.
6. Trevathan E and The Rett syndrome Diagnostic Working Group. Diagnostic criteria for Rett Syndrome. Ann Neurol 1988; 23: 425-8.
7. Hagberg B, Hanefeld F, Percy A, Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol 2002; 6: 293-7.
8. Espinar-Sierra J, Toledano MA, Franco C, Campos- Castello J, Gonzalez-Hidalgo M, Oliete F, Garcia-Nart M. Rett's syndrome: a neurophysiological study. Neurophysiol Clin 1990 Apr; 20(1): 35-42.
9. Kurtoglu S, Atabek ME, Kumandas S, Keskin M. Diabetes mellitus type 1: association with Rett syndrome. Pediatr Int 2005 Feb; 47: 90-1.
10. Hagberg B, Witt-Engerstrom I. Rett syndrome: a suggested staging system for describing impairment profile with increasing age towards adolescence. Am J Med Genet Suppl 1986; 1: 47-59.
11. Hagberg B, Gilbert C. Rett variants: rettoid phenotypes. In: Hagberg B, Anvret M, Wahlstrom J (eds). Rett syndrome: clinical and biological aspects. London: MacKeith Press, 1993, p 40-60.
12. Campos Castelló J. Síndrome de Rett. Rev Neurol (Barc) 1995; 23: 1208-11.
13. Valente KD. Another Rett patient with a typical Angelman EEG. Epilepsia 44: 873-4.
14. Mari F, Azimonti S, Bertani I, et al. CDKL5 Belongs to the Same Molecular Pathway of MeCP2 and it is Responsible for the Early Seizure Variant of Rett Syndrome. Hum Mol Genet 2005; 14: 1935-46
15. Salomao Schwartzman J, Zatz M, dos Reis Vasquez L, et al. Rett syndrome in a boy with a 47, XXY karyotype. Am J Hum Genet 1999; 64: 1781-5.
16. Clayton Smith J, Watson P, Ramsdem S, Black GCM. Somatic mutation in MECP2 as a non fatal neurodevelopmental disorder in males. Lancet 2000; 356: 830-2.
17. Milani D, Pantaleoni C, D'arrigo S, Selicorni A, Riva D. Another Patient With MECP2 Mutation Without Classic Rett Syndrome Phenotype. Pediatr Neurol 2005 May; 32: 355-7.
18. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999; 23: 185-8.
19. Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by methyl-CpG-binding protein MECP2 involves a histone deacetylase complex. Nature 1998; 393: 386-9.
20. Segawa M, Nomura Y. Rett syndrome. Curr Opin Neurol 2005 Apr; 18: 97-104.
21. Neul JL, Zoghbi HY. Rett syndrome: a prototypical neurodevelopmental disorder. Neuroscientist 2004; 10: 118-28.
22. Gibson JH, Williamson SL, Arbuckle S, Christodoulou J. X chromosome inactivation patterns in brain in Rett syndrome: implications for the disease phenotype. Brain Dev 2005 Jun; 27: 266-70.
23. Watson CM, Pelka G, Radziewic T, et al. Reduced proportion of Purkinje cells expressing paternally derived mutant Mecp2308 allele in female mouse cerebellum is not due to a skewed primary pattern of X-inactivation. Hum Mol Genet 2005; 14: 1851-61.
24. Schanen C, Houwink EJ, Dorrani N, et al. Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. Am J Med Genet 2004; 126A: 129-40.
25. Chae JH, Hwang H, Hwang YS, et al. Influence of MECP2 gene mutation and X-chromosome inactivation on the Rett syndrome phenotype. J Child Neurol 2004; 19: 503-8.
26. Smeets E, Terhal P, Casaer P, et al. Rett syndrome in females with CTS hot spot deletions: a disorder profile. Am J Med Genet 2005; 132A: 117-20.
27. Oexle K, Thamm-Mucke B, Mayer T, Tinschert S. Macrocephalic mental retardation associated with a novel C-terminal MECP2 frameshift deletion. Eur J Pediatr 2005 Mar; 164: 154-7.
28. Ballestar E, Ropero S, Alaminos M, et al. The impact of MECP2 mutations in the expression patterns of Rett syndrome patients. Hum Genet 2005 Jan; 116: 91-104.
29. Young JI, Zoghbi HY. X-chromosome inactivation patterns are unbalanced and affect the phenotypic outcome in a mouse model of Rett syndrome. Am J Hum Genet 2004; 74: 511-20.
30. Luikenhuis S, et al. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A 2004; 101: 6033-8.
31. Collins AL, Levenson JM, Vilaythong, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 2004; 13: 2679-89.
32. Jugloff DG, Jung BP, Purushotham D, Logan R, Eubanks JH. Increased dendritic co1mplexity and axonal length in cultured mouse cortical neurons overexpressing methyl- CpG-binding protein MeCP2. Neurobiol Dis 2005 Jun-Jul; 19: 18-27.
33. Armstrong DD. Rett syndrome neuropathology review 2000. Brain Dev 2001 Dec; 23 Suppl 1: S72-6.
34. Thatcher KN, Peddada S, Yasui DH, Lasalle JM. Homologous pairing of 15q11-13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum Mol Genet 2005 Mar 15; 14: 785-97.
35. Battisti C, Formichi P, Tripodi SA, et al. Lymphoblastoid cell lines of Rett syndrome patients exposed to oxidativestress- induced apoptosis. Brain Dev 2004; 26: 384-8.
36. Johnston MV. Clinical disorders of brain plasticity. Brain Dev 2004; 26: 73-80.
37. Mullaney BC, Johnston MV, Blue ME. Developmental expression of methyl- CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience 2004; 123: 939-49.
38. Longo I, Russo L, Meloni I, et al. Three Rett patients with both MECP2 mutation and 15q11-13 rearrangements. Eur J Hum Genet 2004; 12: 682-5.
39. Artigas-Pallares J, Gabau-Vila E, Guitart-Feliubadalo M. El autismo sindrómico: II. Síndromes de base genética asociados a autismo. Rev Neurol 2005 Jan 15; 40 Suppl 1: S151-62.
40. Samaco RC, Hogart A, Lasalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet 2005; 14: 483-92.
41. Makedonski K, Abuhatzira L, Kaufman Y, Razin A, Shemer R. MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum Mol Genet 2005 Apr 15; 14: 1049-58.
42. Ylisaukko-Oja T, Rehnstrom K, Vanhala R, et al. MECP2 mutation analysis in patients with mental retardation. Am J Med Genet 2005; 132: 121-4.
43. Kleefstra T, Yntema HG, Nillesen WM, et al. MECP2 analysis in mentally retarded patients: implications for routine DNA diagnostics. Eur J Hum Genet 2004; 12: 24-8.
44. Hitchins MP, Rickard S, Dhalla F, et al. Investigation of UBE3A and MECP2 in Angelman syndrome (AS) and patients with features of AS. Am J Med Genet 2004; 125A: 167-72.
45. Borg I, Freude K, Kubart S, et al. Disruption of Netrin G1 by a balanced chromosome translocation in a girl with Rett syndrome. Eur J Hum Genet 2005, 13: 921-7.
46. Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autismspectrum disorders. Hum Mol Genet 2004; 13: 629-39.
47. Campos-Castelló J, De Yébenes JG, Mena MA, Oliete García F. Cerebrospinal fluid concentration of Homovanillic Acid and 5-Hydroxindoleacetic Acid in patients with Rett Syndrome. International Pediatrics 1991; 6: 340-2.
48. Horike S, Cai S, MiyanoM, et al. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet 2005; 37: 31-40.
49. Goyal M, O'Riordan MA, Wiznitzer M. Effect of topiramate on seizures and respiratory dysrhythmia in Rett syndrome. J Child Neurol 2004; 19: 588-91.
50. Giampietro PF, Schowalter DB, Merchant S, Campbell LR, Swink T, Roa BB. Widened clinical spectrum of the Q128P MECP2 mutation in Rett syndrome. Child's Nerv Syst 2006; 3: 320-4.
51. Kerr AM, Webb P, Prescott RJ, Milne Y. Results of surgery for scoliosis in Rett syndrome. J Child Neurol 2003, 18: 703-8.
52. Ramaekers VT, Blau N. Cerebral folate deficiency. Dev Med Child Neurol 2004; 46: 843-51.
53. Lotan M, Isakov E, Merrick J. Improving functional skills and physical fitness in children with Rett syndrome. J Intellect Disabil Res 2004; 48: 730-5.
54. Yasuhara A, Sugiyama Y. Music therapy for children with Rett syndrome. Brain Dev 2001, 23 (suppl 1): S82-4.
55. Bumin G, Uyanik M, Kayihan H, Duger T, Topcu M. The effect of hand splints on stereotipies hand behavior in Rett syndrome. Turk J Pediatr 2002; 44: 25-9.
56. Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007; 315: 1143-7.

