










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.68 n.3 Ciudad Autónoma de Buenos Aires mayo/jun. 2008
Diagnosis and treatment of congenital hemophilia with inhibitors. A Latin American perspective
Raúl Pérez Biancoa, Margareth Castro Ozelob, Paula Ribeiro Villaçac, María Helena Solanod, Guillermo Jiménez Cruze, Carlos Martinez Murillof, Jaime García Chavezg, Saul Mendozah , Ismael Rodríguez Greccoi, Arlette Ruiz-Saezj
aInstituto de Investigaciones Hematológicas, Academia Nacional de Medicina, Fundación Nacional de Hemofilia, Buenos Aires, Argentina;
bHemocentro de Campinas, Universidade Estadual de Campinas, Campinas, Brazil;
cCentro de Hemofilia, Hospital das Clínicas, Universidade de São Paulo, São Paulo, Brazil;
dHospital de San Jose, Fundación Universitaria Ciencias de la Salud, Bogotá, Colombia;
eHospital Mexico, Caja Costarricense de Seguro Social, San José, Costa Rica;
fUnidad Médica de Alta Especialidad, Centro Médico Nacional Siglo XXI, Ciudad de México, México;
gUnidad Médica de Alta Especialidad, Centro Médico Nacional La Raza, Ciudad de México, México;
hHospital Nacional Edgardo Rebagliati Martins - EsSalud, Lima, Peru;
iServicio de Medicina Transfusional, Centro Hospitalario Pereira Rossell, Montevideo, Uruguay;
jCentro Nacional de Hemofilia, Banco Metropolitano de Sangre, Caracas, Venezuela
Postal address: Dr. Raúl Pérez Bianco, Instituto de Investigaciones Hematológicas, Academia Nacional de Medicina, J.A. Pacheco de Melo 3081, 1425, Buenos Aires, Argentina. Fax: (54-11) 4805-0712 e-mail: perezbianco@hematologia.anm.edu.ar
Summary
The Committee of Latin America on the Therapeutics of Inhibitor Groups (CLOTTING) is composed of a number of hemophilia specialists from Latin America. The group aims to encourage the adoption of a good standard of care for Latin American patients with hemophilia. The occurrence of inhibitors in patients with hemophilia poses clinical challenges, and it is estimated that between 1000 and 3 000 patients in Latin America are affected by hemophilia with inhibitors. There is an urgent need to establish a regional consensus and clinical guidelines for the diagnosis and treatment of these patients. We present an extensive review based on best current clinical practice and published literature, as seen from a Latin American perspective, taking into account the variable nature of hemophilia care available in the various countries in this Region.
Key words: Hemophilia; Inhibitors; Treatment; Hemophilia guidelines
Resumen
Diagnóstico y tratamiento de la hemofilia congénita con inhibidores. Una perspectiva latinomericana. El Comité Latinoamericano sobre la Terapéutica de Personas con Inhibidores (CLOTTING) está compuesto por un grupo de especialistas en hemofilia de Latinoamérica. El objetivo del grupo es promover la adopción de un estándar de tratamiento óptimo para los pacientes con hemofilia en Latinoamérica. La prevalencia de inhibidores en pacientes con hemofilia en Latinoamérica determina desafíos clínicos y se estima que de 1000 a 3000 pacientes en esta región están afectados con hemofilia e inhibidores. Existe una necesidad urgente de establecer un consenso regional y guías clínicas para el diagnóstico y tratamiento de estos pacientes. Nosotros presentamos una revisión exhaustiva basada en las mejores prácticas clínicas vigentes y en los datos publicados en la literatura, con una perspectiva latinoamericana, tomando en cuenta la variabilidad existente de los tratamientos de la hemofilia disponibles en los diferentes países de esta Región.
Palabras clave: Hemofilia; Inhibidores; Tratamiento; Guías clínicas
Hemophilia is an inherited bleeding disorder characterised by frequent bleeding episodes, particularly affecting joints and muscles. Treatment with clotting factors can control the bleeding, preventing chronic arthropathy and premature death. However, in as many as 20-30% of patients with hemophilia A (who have a deficiency of factor VIII [FVIII]), and 3% of patients with hemophilia B (who have a deficiency of factor IX [FIX]), replacement therapy with coagulation factors is associated with the development of inhibitors1, 2.
Inhibitors are neutralising antibodies that interfere with the function of FVIII or FIX. The occurrence of inhibitors has significant clinical implications (the response to treatment becomes uncertain, morbidity is increased and life expectancy is reduced). In addition, direct medical costs are much higher for hemophilia patients who have inhibitors, as are the non-medical costs to the patients, their families and society3.
Latin America (LA) has a population of around 500 million. The population is heterogeneous, and ethnic mixtures predominate in many countries. According to the World Federation of Hemophilia Global Survey, in 2006 there were 20,583 hemophilia patients registered in LA4. It is estimated that between 1,000 and 3,000 hemophilia patients in LA have inhibitors. This estimate is based on the inhibitor prevalence reported in different Latin American countries - at between 11% and 19% -the reported prevalence is similar to that described for other populations5-9 .
It is recommended that national hemophilia programmes be developed; these must consider a range of issues including: organisation and infrastructure; appropriate diagnosis; aims of treatment; methods of treatment; and the products that are available10. However, many Latin American countries do not have established national hemophilia programmes. Indeed, mainly due to economic reasons, the treatment of hemophilia is not a priority in these countries. As a result, the level of hemophilia care varies widely, and a homogeneous or standardised approach is not apparent even within individual countries; this is particularly evident with regard to inhibitor diagnosis and treatment. Consequently, there is an urgent need to establish a consensus as to how to best manage these patients. This review was made with the purpose of facilitating the implementation of best practice in hemophilia and inhibitor treatment.
The CLOTTING group
The Committee of Latin America on the Therapeutics of Inhibitor Groups (CLOTTING) is composed of a number of hemophilia specialists from LA. Group members are listed in Appendix B. The objective of the group is to discuss and review the diagnosis and treatment of inhibitor patients in LA. The group also supports the adoption of best practice in hemophilia and inhibitor treatment, communicating and encouraging the improvement of hemophilia care in the region through a constant exchange of experiences.
A total of eight CLOTTING meetings have taken place to prepare this review on the diagnosis and treatment of patients with congenital hemophilia with inhibitors.
1. Diagnosis of inhibitors
1A. Inhibitor surveillance in hemophilia
According to literature data available and considering the cost of reagents and other budget constraints in Latin American countries, in patients with hemophilia, inhibitor screening should take place:
-In severe and moderately severe hemophilia A, previously untreated patients should be screened for inhibitors after every 5th exposition day (ED) until the 20th ED., then 3 to 6 monthly up to 150 ED, then once every 12 months (grade C recommendation based on level IV evidence)11.
-In mild hemophilia A, screening of inhibitors is recommended after intensive replacement therapy, especially in individuals with high risk mutations (grade B level III)11.
-Prior to any surgical or invasive procedure.
-When the clinical (or laboratory) response to conventional replacement therapy is poor, or the frequency of bleeding increases11.
-In severe and moderate hemophilia B, the frequency of screening for inhibitors should be the same as for severe hemophilia A (grade B recommendation based on level III evidence)11.
-Following prolonged treatment with continuous infusion (CI) or a high-dose therapeutic regimen.
Inhibitor screening is usually performed using activated partial thromboplastin time (aPTT) mixing/incubation testing. As FVIII inactivation by inhibitors is time and temperature dependent, the aPTT of the patient: pooled plasma mixture should be measured and compared immediately after mixing and after a 2-hour incubation period12.
1B. Inhibitor detection and quantification
The Bethesda assay is the recommended standard method for measuring FVIII inhibitor titre. The same method may also be used to quantify FIX inhibitors, but without the 2-hour incubation (grade C recommendation based on level IV of evidence)12.
The Nijmegen modification of the Bethesda assay can avoid false-positive results. This is now recommended by the International Society of Thrombosis and Haemostasis (ISTH) Factor VIII/IX Scientific Subcommittee13-15. However, as the procedure can pose an additional burden when performing laboratory tests, Nijmegen modification is sometimes considered optional; it is made only when resources are available and at physicians' discretion.
In LA, the most important parameter evaluated with regard to low-titre inhibitors is patients' clinical response. However, data relating to factor recovery and plasma half-life may be required for the detection of low-level inhibitors that reduce factor survival11. They may be performed in selected cases, when resources are available, and also in special circumstances, such as in the evaluation of immune tolerance regimens.
1C. Classification of inhibitors
The ISTH classification of inhibitors16 is accepted worldwide. Low responders have inhibitor levels <5 Bethesda Units (BU) ml-1 and do not develop an increase in inhibitor levels after further exposure to FVIII/FIX. High responders have inhibitor titres >5 BU ml-1 or, if levels have decreased below this, titres that will rise to >5 BU ml-1 on re-exposure to FVIII/FIX.
1D. Standardization and quality control
Laboratories diagnosing hemophilia and inhibitors should standardise FVIII and FIX assays. The World Federation of Hemophilia (WFH) laboratory manual17, or the ISTH Factor VIII/IX Scientific Subcommittee recommendations [available from the ISTH website]18 are valuable tools for this purpose. An external quality assessment using World Health Organization (WHO) coagulation standards through the International External Quality Assessment Scheme (IEQAS) of the WFH Laboratory Quality Control Programme19 are also advisable in the quality control of coagulation laboratories.
1E. Genetic diagnosis
Although some genetic defects are associated with inhibitor development20, molecular studies for the detection of FVIII or FIX gene mutations are not routinely carried out in LA. Few centres in the region have the capability to conduct genetic investigations on inhibitor patients. More co-operative studies performed between hemophilia centres in LA are needed to establish a reference database of common gene defects associated with inhibitors in the LA population. CLOTTING group members' centres that can perform such studies are marked with an asterisk (*) in Appendix B. Up to now, few genetic investigations have been made in the Latin American population21-23.
2. Classification of bleeding episodes
After reviewing different definitions of bleeding severity in the literature, bleeds occurring in patients with hemophilia can be classified into two categories24, 25.
Mild and moderate bleeding
In mild bleeding, signs and symptoms of hemorrhage are evident but these do not prevent patients from performing normal activities. Patients experiencing moderate bleeds exhibit signs and symptoms of hemorrhage and are prevented from performing normal activities.
Examples of mild and moderate bleeding include: nose and gum bleeds without hemodynamic repercussions; soft-tissue bleeds and superficial skin cuts; acute hemarthroses; peripheral muscular hematomas; hematuria; and bleeding without hemodynamic repercussions associated with dental extraction.
Severe bleeding
This is defined as any bleeding that poses a risk to life, limb, or an important function (life- or limb-threatening hemorrhages). Examples include: bleeding associated with the central nervous system, thoraco-abdominal and retroperitoneal bleeds; gastrointestinal bleeds; neck and throat bleeds; large hemarthroses and large muscular hematomas (iliopsoas hemorrhage or bleeding causing compartment syndrome); opthalmic (intra-ocular) hemorrhages; severe trauma; and any external/internal bleeding with hemodynamic repercussions. Any bleeding in hemophiliac patients, particularly patients with inhibitors, could be severe (life- or limb-threatening) if it is not immediately treated.
3. Treatment of bleeding episodes in congenital hemophilia A with inhibitors
The management of acute bleeds in hemophilia A patients with inhibitors depends on the severity of the bleed, the inhibitor titre at the time of bleeding and any known anamnestic response. When choosing a treatment option, the efficacy, safety and availability of the therapy should also be considered. Hemostatic treatment should be initiated as soon as possible.
3A. Mild and moderate bleeding episodes
Due to a limited number of randomised clinical trials evaluating treatment options for inhibitor patients in different clinical settings, a review of current management of bleeding in patients with congenital hemophilia A and inhibitors is made available in the tables below.
3Ai. Low responders (Table 1)11, 26-28
TABLA 1.- Treatment of mild and moderate bleeding in hemophilia A patients with low-response inhibitors

3Aii. High responders11, 26-38
Recombinant activated factor VII (rFVIIa; NovoSeven©, Novo Nordisk, Bagsvaerd, Denmark) and activated prothrombin complex concentrate (aPCC; FEIBA©, Baxter, Deerfield, IL, USA) are the preferred bypassing agents for mild and moderate bleeds in high-responder patients. Efficacy rates with these agents are around 79-92%29-32. A recent head-to-head comparison between both treatments has just been published showing that aPCC and rFVIIa appear to have a similar effect on joint bleeds although the efficacy between products is rated differently by a substantial proportion of patients33. However, Young et al demonstrated that rFVIIa 270 µg/kg and rFVIIa 90 µg/kg (x3 doses) reduces the need for rescue medication compared to aPCC suggesting it to be potentially more effective treatment option than aPCC for home treatment joint bleeds34. In addition, Ozelo et al, demonstrated that rFVIIa resolve bleeds more quickly (4.4 hs.) than aPCC (62.6 hs.) and was more effective (100% vs. 57.7% respectively)35. Megadoses of rFVIIa (doses >200 µg kg-1body weight) are being used in inhibitor patients for mild and moderate bleeding as a convenient means of avoiding repeated injections, particularly in children. Although this is a non-approved use, it has been extensively tested without any safety concerns34, 35. In a recent trial, comparing three standard doses of rFVIIa 90 µg kg-1 body weight every 3 hours with a single dose of rFVIIa 270 µg kg-1 in the home setting, administration of the single high dose of rFVIIa had comparable efficacy and safety to standard dosing, and no thromboembolic events were reported (Table 2)34, 36, 37. When these options are not available, non-activated prothrombin complex concentrates (PCCs) can be used. The usual dose of these is 50-75 U kg-1 body weight every 12 hours. Efficacy rates in randomised clinical trials performed in the early 1980s were around 50%27, 40, 41.
TABLA 2.- Treatment of mild and moderate bleeding in hemophilia A patients with high-response inhibitors

3B. Severe bleeding episodes
In instances of severe bleeding, the duration of treatment must be considered for each case on an individual basis and therapy should subsequently be evaluated according to the clinical response. In addition, prophylaxis scheme should be consider according to the type and severity of bleeding42.
3Bi. Low responders and high responders with an initially low titre (<5 BU ml-1)14, 26-28
It has been recommended to maintain FVIII:C levels >30 IU dl-1 when using high doses of FVIII as the first option for low-responder or high-responder patients with an initially low titre27. When choosing high doses of FVIII or aPCC as the first option for high-responder inhibitor patients who have a low titre at the beginning of treatment, the risk of developing an anamnestic response must be considered (Table 3)27, 28, 32.
TABLA 3.- Treatment of severe bleeding in hemophilia A patients with low-response inhibitors or high-response inhibitors with an initially low titre

3Bii. High responders (Table 4)14, 26-38, 42, 43
TABLA 4.- Treatment of severe bleeding in hemophilia A patients with high-response inhibitors

The concomitant use of antifibrinolytics is discussed in Section 9A.
4. Treatment of bleeding episodes in congenital hemophilia B with inhibitors
Treatment options for patients with congenital hemophilia B and inhibitors will depend on the severity of the bleed and the past history of anamnestic response. Patients with hemophilia B and inhibitors can exhibit allergic reactions following FIX administration; this is particularly possible in patients with large gene deletions44. The allergic reaction includes urticaria, rash, angio-oedema, bronchospasm, hypotension or acute anaphylaxis. The reactions can appear at the same time as the inhibitor (median of 11 days of exposure), and after infusion of both high- and low-purity FIX concentrates14. An association between allergic reactions and nephrotic syndrome has been reported during immune tolerance induction with FIX44-46. Consequently, the possibility of patients experiencing anaphylaxis or other allergic reactions should be considered when selecting FIX-containing products.
4A. Patients without a history of allergic reactions
Low-titre inhibitor patients may respond to high-purity FIX. For mild and moderate bleeds, the initial recommended dose is 100-200 IU kg-1 body weight, followed by 50-100 IU kg-1 body weight every 12 or 24 hours for as long as is judged necessary to control the bleeding (usually 1-3 days). In cases of inadequate response, treatment should be changed to bypassing agents.
Severe bleeds or high-responder patients should be treated with bypassing agents using doses similar to those described for patients with FVIII inhibitors (Tables 2 and 4). The risk of anamnesis and anaphylaxis should be borne in mind when choosing aPCC to treat patients with hemophilia B and inhibitors. rFVIIa might be considered the treatment of choice for acute bleeds14, 28.
4B. Patients with a history of allergic reactions
Because of the possible recurrence of allergic reactions following FIX administration, rFVIIa is recommended as the treatment of choice in this group of patients14, 28. Doses are similar to those used for hemophilia A patients with inhibitors. If the requirement for treatment is urgent and rFVIIa is not available, FIX-containing products may be used with extreme caution under medical supervision. Premedication with antihistamines and steroids is recommended, together with adequate conditions for the treatment of acute allergic reactions and anaphylaxis14.
5. Home treatment for mild and moderate bleeds
Early studies demonstrated the significant quality-of-life benefits associated with home treatment47-50. Home infusion of coagulation factors significantly decreased hospital admissions due to bleeding complications; it also decreased pain, dysfunction and long-term disability47-52. In patients with hemophilia and inhibitors, home treatment of mild and moderate bleeds has shown results similar to those found in patients without inhibitors29-32.
While the introduction of home treatment undoubtedly motivates patients and contributes positively to quality of life, properly performed self-management also puts demands on patients in terms of observation, judgement and skilled reporting. To meet these demands, patients should comply with recommendations and co-operate with treatment centre staff. Ideally, patients should be alert to the onset of new bleeding episodes and capable of taking action immediately. When selected for this modality, patients should be taught to understand the properties of products prescribed. Even for highly skilled patients, complete training and instructions should be given concerning product storage, product reconstitution and a practical approach to self-injection.
rFVIIa/aPCC home-treatment programmes require the education of patients and their families in relation to dose, administration frequency and evaluation of response. Such programmes must be designed and closely monitored by hemophilia treatment centres. Home-treatment training should be continuously validated by hemophilia centre staff. It is particularly important that patients understand product characteristics, especially the short half-life of rFVIIa and the requirement for repeated dosing at regular intervals when appropriate, so that additional dosing is not unduly delayed53. Patients should contact their treatment centres for advice in any cases of doubt or if treatment appears to be ineffective.
It is recommended that patients make a registry of all bleeding episodes treated at home. The bleeding site, time of onset, treatment used and duration should be properly recorded by patients or their caregivers and communicated to the hemophilia centres.
Hemophilia centres should be capable of supplying sufficient quantities of product to completely treat mild and moderate bleeding and to begin the treatment of severe bleeding in cases of emergency. They should also provide patients with appropriate advice and support on a 24-hour basis. Measures should be taken to ensure the proper running of home-treatment programmes, with adequate stocks and easy delivery of concentrates being required to realise the full benefits of this treatment modality53.
Infusion sets (syringes, needles and vials) should be properly discarded as they contain biological material. Patients should place items in appropriate, safe, disposable boxes, if possible provided by the hemophilia centre. If safe disposal by patients cannot be assured, it is recommended that material is returned to hemophilia centres for adequate disposal.
Like in non-inhibitor patients, treatment should start as early as possible, preferably less than 2 hours after symptoms become apparent.
Based on the review made, appropriate dosage recommendations for the treatment of mild and moderate bleeds are given in Tables 1 and 2. According to the treatment response, 1-4 doses are generally required. However, if an effective response is not apparent after the first two doses, patients should contact their treatment centres for instructions.
6. Prophylaxis for patients with congenital hemophilia and inhibitors
Currently, there is no clinical evidence in the literature to determine whether prophylaxis is more effective than on-demand treatment for hemophilia patients with inhibitors. Attempts to establish a prophylactic regimen for inhibitor patients with frequent bleeds (secondary prophylaxis) have been carried out using bypassing agents (both rFVIIa and aPCC) with the objective of decreasing the number of recurrent bleeds and allowing the rehabilitation of target joints. Further information is required with regard to minimum effective doses, dosing frequency, and safety aspects relating to prolonged use of these agents in prophylaxis54-59.
6A. Prophylaxis with rFVIIa
Isolated case reports using rFVIIa as prophylaxis in high-responder inhibitor patients have been described using different doses (ranging from 90 µg kg-1 body weight daily, to 200 µg kg-1 body weight every 6 hours for prolonged periods), with variable results. A multi-centre, randomized, prospective trial evaluating the efficacy and safety of rFVIIa for secondary prophylaxis in inhibitor patients has just been finalized. It showed that rFVIIa dosed once-daily in inhibitor patients with frequent bleeds, either with 90 µg kg-1 body weight or 270 µg kg-1 body weight, resulted in clinically relevant reductions in number of bleeds and improvements in quality of life during prophylaxis compared with conventional on-demand therapy, without raising any safety concerns54-56. Hoots et al, recently published the results of a prospective trial showed that secondary prophylaxis with rFVIIa may provide an alternative treatment modality to conventional treatment in patients with frequent bleeds related to health-related quality of life (HRQoL) improvement57.
6B. Prophylaxis with aPCC
Retrospective studies and case reports have described the prophylactic use of aPCC, particularly for late postsurgical or severe bleeding episodes, for physical therapy or rehabilitation, and to prevent recurrent haemarthroses in target joints, e.g. during immune tolerance induction. Doses are similar to those recommended for bleeding episodes (50-100 U kg-1 body weight), administered three times a week, or every other day. Results have shown a reduction of up to 50% in the number of joint bleeds, together with the prevention of severe hemorrhages58, 59. Leissinger et al, described 5 patients under prophylaxis, dosages ranged from 50 to 75 U kg-1 three times a week in four patients and 100 U kg-1 daily in one patient. This short case series suggest thats prophylaxis with aPCC may be an option for decreasing the number of bleeding episodes60.
7. Surgery in congenital hemophilia with inhibitors
Surgery in patients with hemophilia and inhibitors is always a high-risk procedure. It should be carried out at experienced centres by skilled surgeons, under the supervision of a hematologist. Involvement of a multidisciplinary team is required both before and after the surgery. Hemostasis must be adequate throughout the entire procedure, and for a period of days afterwards to allow wound healing. Elective surgery requires excellent pre-surgical evaluation and appropriate information, both for patients and their families. Enough factor concentrate should be available to cover the entire treatment and any possible ensuing complications. It is also important that a hemostasis laboratory be involved in determining the appropriate dosage of coagulation factor, and the detection and quantification of inhibitors. Pharmacokinetic (PK) studies, for both adults and children, are not routinely performed in inhibitor patients in the surgical setting. However, PK information can be useful when performing surgery in low-responder patients. When conducting surgery under rFVIIa coverage, it may also be appropriate to perform PK evaluations before the surgery, as individual responses, namely half-life and clearance, are variable, particularly in younger patients61, 62.
Two types of surgical interventions have been reported in the literature - minor and major. However, no clear definition of these has been published to date63. The differentiation between major and minor procedures is based on the extent of replacement material necessary for fibroblast growth and good scar formation64. Minor surgery may be considered to involve procedures that do not penetrate a body cavity or transect a major limb bone, and major surgery may be regarded as any type of thoracoabdominal, intracranial or orthopedic surgery65, 66. Based on the available literature, the classification of minor surgery should include only those procedures requiring small skin excisions and/or small sutures66. All the other procedures should be classified as major surgery. Among orthopedic procedures, only synoviorthesis and arthrocentesis are classified as minor63-66.
There has been no agreement to evaluate efficacy parameters relating to the hemostatic response to substitution therapy after surgical procedures. Effective hemostasis is generally assessed in terms of physicians' clinical judgement. A clinical trial of surgery in inhibitor patients used the following definitions67:
- Treatment is rated as effective if bleeding is prevented during surgical intervention.
- Treatment is rated as partially effective if the bleeding is only partially prevented during surgical intervention.
- Treatment is rated as ineffective if bleeding cannot be prevented during surgical intervention.
Recent literature reviews have suggested that, with the options available, it is possible to achieve adequate hemostasis, with a very low rate of side effects, during elective surgery in patients with inhibitors68, 69. Both rFVIIa and aPCC have demonstrated efficacy rates between 80% and 90%, or even higher, although optimal doses and dosing intervals have not been clearly established69-72. Ingerslev et al and Obergfell et al reviewed the data on elective orthopedic surgeries in hemophilia patients with inhibitors under cover of rFVIIa. These authors highlight the relevance of rFVIIa in elective orthopedic surgery regarding the safety and efficacy according to the published experience particularly in major procedures73, 74.
It is extremely important to have enough factor concentrate to cover the entire treatment and to avoid complications. If possible, it is recommended to have both options available before deciding on major elective surgery.
Porcine FVIII was not included in this review because it has been recently discontinued and is no longer available in the market. A recombinant porcine factor VIII is being investigated in clinical trials.
In this review, recommendations for surgery in patients with congenital hemophilia and inhibitors are similar to those for severe bleeding (Tables 3 and 4) with some important qualifications:
i. The inhibitor should be quantified prior to any surgical procedure.
ii. Preferably, elective surgery in inhibitor patients should be scheduled for the beginning of the week and not be carried out before weekends or holidays.
iii. When choosing high doses of FVIII or aPCC as a first option for the high-responder inhibitor patient with a low titre at the beginning of treatment, the risk of developing an anamnestic response must be considered27, 28, 32.
iv. Initial (pre-operative) dosing should be performed immediately before the beginning of surgery, preferably in the operating room during induction anaesthesia.
v. It has been recommended to maintain FVIII:C levels >50 IU dl-1 when using high doses of FVIII as a first option for low-responder or high-responder hemophilia A patients with an initially low inhibitor titre27.
vi. As there is not enough evidence to recommend the use of high doses of FIX in surgery for low-responder or high-responder hemophilia B patients with an initially low inhibitor titre, bypassing agents are recommended.
vii. In surgery for hemophilia patients, sutures are preferred over cauterization of small vessels.
viii. The duration of treatment depends on the type of surgery. In general, treatment periods of 1-5 days for minor surgery and 7-14 days for major surgery are adequate.
ix. In cases of bleeding, or if treatment is partially effective, a surgical cause should be excluded. If there is persistent bleeding despite control of a surgical cause, the treatment should be modified by increasing the dose or changing the treatment option.
x. Central venous access devices (CVAD) are often used for hemophilia patients with inhibitors under immune tolerance therapy (ITT). Although external catheters such as the Hickman© and Broviac© (Bard Access Systems, Inc., Salt Lake City, Utah, USA), have frequently been used for ITT in hemophilia patients, there is a growing evidence that inhibitor patients with fully implantable catheters (portacaths) have fewer catheter-related infections. This suggests that ports may also be preferable, unless the advantages of external catheters are important for particular individuals according to physicians' judgement75. Several haemostatic coverage regimens at the time of CVAD placement and removal have been described. Effective replacement treatment during CVAD insertion was generally obtained using rFVIIa or aPCC. The usual the duration of treatment is 2-3 days76-78.
xi. The management of dental procedures is subject to the same recommendations relating to the type of anesthesia used and local measures, for both hemophilia patients with and without inhibitors. Replacement therapy is recommended whenever general anesthesia with endotracheal intubation or nerve trunk infiltration is going to be performed. Truncal anesthesia carries the risk of life-threatening cervical bleeding for inhibitor patients. A careful approach by a skilled dentist and more prolonged factor replacement than a single pre-procedure dose is recommended. For dental extraction, the same replacement therapy as for a minor surgery is recommended, although this is normally necessary for only 24-48 hours. Local hemostatic measures, such as adequate suturing, use of fibrin glue and use of systemic antifibrinolytic agents, if not contraindicated, should always be employed.
xii. When invasive diagnostic procedures (such as lumbar puncture, arterial puncture, bronchoscopy with brushing or biopsy, or gastrointestinal endoscopy with biopsy) are performed in inhibitor patients, replacement therapy with at least a single dose of coagulation factor immediately prior to the procedure is recommended. However, considering the high risk of serious complications in these procedures, especially lumbar punctures, 2-3 additional doses after the procedure can be necessary.
A consensus on orthopedic surgery in inhibitor patients was published by a panel of experts63. The recommendations (shown in the adapted table below) were made in order to obtain effective hemostasis during these procedures and can be used as another option for surgery in patients with inhibitors (Table 5). Occasionally, unsatisfactory results have been observed with conventional doses of bypassing agents, particularly in orthopedic surgery. Based on both published case series and personal experience, some authors have described the use of higher than recommended doses of rFVIIa and aPCC32, 72, 79.
TABLE 5.- Recommended dosage of recombinant activated factor VII (rFVIIa) and activated prothrombin complex concentrate (aPCC) for surgery (adapted from data taken from Rodriguez-Merchan et al.)63

8. Immune tolerance induction (ITI)
ITI is a strategy to desensitise patients' immune systems to extrinsic FVIII or FIX by repeated intensive exposure to this antigen. This approach is successful in eliminating the inhibitor permanently in around 80% of inhibitor patients. ITI may take 1-3 years to achieve tolerance, at a cost of approximately 1 million US$ per patient80. ITI is recommended for high-responder inhibitor patients.
Although it is almost 30 years since the first experience of the induction of tolerance (at Bonn, Germany)81, the mode of action of ITI is still not fully understood, and little is known about the mechanism by which immune tolerance is achieved. Future immunologically based studies are needed to clarify this. Various mechanisms have been proposed to explain the effect of repeated intensive exposure of FVIII or FIX on immune system. Down-regulation of the immune response to factor VIII and long-lasting tolerance could be induced by anti-idiotypic antibodies neutralising anti-FVIII antibodies, the deletion of antigen-specific T cell clones or inducing the development of regulatory T-suppressor cells82.
Protocols combining intensive FVIII or FIX replacement with concomitant immunosuppression exist. The Malmö regimen, in which high-dose FVIII or FIX replacement is combined with cyclophosphamide, high-dose immunoglobulin and protein A immuno-adsortion, is the best-known example of these83. The use of immunosuppressive agents such as the anti-CD 20 monoclonal antibody, rituximab, has also been reported in the treatment of inhibitor patients84-87.
Regimens of ITI are the only available treatments that eradicate alloantibodies to FVIII or FIX, and these remain the best long-term option, particularly for high-titre antibodies88. Inhibitor eradication with ITI leads to a marked improvement in patients' quality of life and may considerably reduce the cost of future treatment.
Data relating to ITI have been collated in several registries. These include the International Immune Tolerance Registry (IITR)89 the North American Immune Tolerance Registry (NAITR)90, the German91 and the Spanish92 registries. The overall success rate of ITI in all these registries is comparable, at 75 ± 10%. Favourable indicators and adverse risk factors relating to outcomes in ITI have been identified using the accumulated data. Recently, Di Michele et al, published a consensus recommendation on immune tolerance induction based on the level of available supporting evidence93.
There is agreement that a low-inhibitor titre prior to the initiation of ITI (<10 BU ml-1) and a low historical peak inhibitor titre (<200 BU ml-1) are favourable clinical features. However, there is some disagreement as to whether patient age at the initiation of ITI, or the time between inhibitor diagnosis and starting ITI, predicts successful outcome (Table 6).
TABLE 6.- Immune tolerance induction - influence of factor VIII (FVIII) dose or immunosuppression on success rate [adapted from data taken from DiMichele 200290 and DiMichele 200388 and DiMichele 200793]
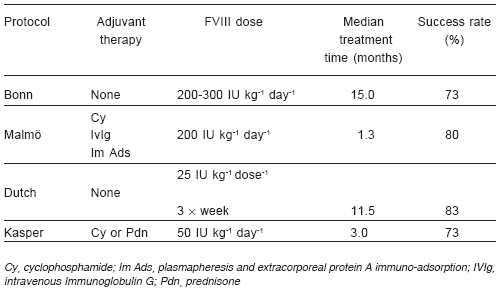
The study cohorts, as well as the immune tolerance regimens, differ in many respects, including the dosage of FVIII, the number of infusions of factor concentrate per day, the type of factor concentrate, the association with immunosuppressive drugs and the definition of success of ITI82. The influence of the type of product used has not been well established. Although Kreuz and colleagues have suggested that successful immune tolerance may be achieved using intermediate-purity FVIII concentrate94, recent results suggest good efficacy may be obtained using high-purity products or recombinant FVIII (rFVIII)95, 96. Success rates in these studies are similar, although the median time to success differs.
Low-dose ITI is a less-demanding protocol in which patients have to be infused only two or three times a week. As the amount of FVIII infused is lower, such a regimen may be initially more attractive for economic reasons. However, one disadvantage as compared to high-dose ITI regimens, is that patients may need to be treated for longer periods of time before tolerance is achieved97.
ITI has been carried out in LA with either a modified high-dose protocol98, 99 or the low-dose Dutch protocol using FVIII/von Willebrand factor (VWF) concentrate (30 IU kg-1 body weight three times weekly)100. Results are similar to those published in the literature, and co-operative studies in the region are encouraged.
A central venous catheter is usually used when ITI is carried out, particularly in children whose regimens require daily, or several weekly, doses. Infection in this line is a common complication and is related to an anamnestic increase in inhibitor titre93, 96.
Relapses following successful ITI are reported, but they appear to be infrequent. The International Registry reports 6 relapses in 128 patients in whom ITI was successful. The relapse rate was 15% at 15 years101.
Experience with ITI for hemophilia B patients with inhibitors is limited. The principles of treatment for these patients are similar to those for hemophilia A. However, the success rate is lower, regimens are associated with a risk of anaphylaxis to FIX, and irreversible nephrotic syndrome can occur46, 102. Recently, DiMichele et al, concluded that there can be no recommendations for ITI in hemophilia B93.
9. Special considerations
9A. Use of antifibrinolytics
Concomitant use of antifibrinolytics and bypassing agents during bleeding or surgery has not been investigated in clinical trials. However, some authors have suggested that the use of systemic antifibrinolytics, particularly tranexamic acid, together with rFVIIa is safe, and can help to prevent bleeding30, 71, 103, 104. This could be recommended for mucosal bleeding (with a mouthwash being a possibility here) and orthopedic surgery, unless there is a known contraindication such as urinary bleeding or suspected disseminated intravascular coagulation. Concurrent systemic antifibrinolyitic therapy with aPCC is generally avoided, and there is no evidence relating to the safety of this. At least a 12-hour interval between the last dose of aPCC and the use of systemic antifibrinolytics should be considered.
Recommended antifibrinolytic doses:
-Tranexamic acid: 25 mg kg-1 body weight every 8 hours PO or 15 mg kg-1 body weight every 8 hours IV; mouth wash at 10% solution.
-Epsilon amino caproic acid: 50-100 mg kg-1 body weight every 6 hours PO or IV (maximum dose is 24 g day-1).
9B. Continuous infusion (CI) of rFVIIa
Although some evidence of the efficacy rFVIIa administered by CI has been published, more studies are needed to allow the recommendation of this treatment modality.
As for FVIII and FIX, the rationale behind CI with rFVIIa is a consequence of its convenience (because its short half-life and requirement for frequent dosing) and the potential cost-effectiveness associated with a reduced total dose103-105. Although FVII:C levels attained in plasma are not always predictive of efficacy, surgical efficacy rates greater than 90% have been obtained with an initial bolus dose of 90-120 µg kg-1 body weight, followed by CI of 10-50 µg kg-1 h-1, aiming at a FVII:C level of 20-30 IU ml-1 106-109. However, cost advantages have still to be demonstrated, particularly when additional bolus doses are required to control bleeding109.
9C. Laboratory monitoring
Currently, there is no measurable laboratory parameter for monitoring the efficacy of bypassing agents. Assays using non-flow techniques, like prothrombin time (PT) and aPTT, have limited application in the monitoring of these agents and do not reflect physiological conditions110. For rFVIIa, using either bolus dosing or CI, FVII:C levels do not generally correlate closely with the clinical response107. Experimental models using whole blood, and indirectly measuring thrombin generation, may have a role in the monitoring of bypassing agents107. Young and co-workers, suggested that thromboelastography may potentially be a clinically useful tool for monitoring changing concentrations of recombinant activated factor VII in hemophilia patients, but only when the baseline curve is significantly abnormal111. On the other hand, Lak et al, showed that ROTEM© should not be the method of choice for monitoring rFVIIa therapy in Glanzmann patients112.
Investigations are being carried out using thromboelastography and thrombin generation assays (TGA), but standardization and validation is currently lacking113, 114. Treatment efficacy during bleeding and surgery is usually judged clinically.
9D. Adverse events with hemostatic drugs
Antifibrinolytics: These drugs inhibit plasmin generation and activity. Adverse events include: allergic reactions; nausea; diarrhoea; hypotension; and fatigue. It is necessary to be wary of the use of these drugs in patients with kidney failure, haematuria, cardiopathy or liver disease. Rapid intravenous administration must be avoided as this can result in hypotension and cardiac arrhythmias.
aPCCs: As these products are obtained from human plasma, the risk of transmission of infectious agents should be considered. However, for known human pathogens, this risk is very low. Mild or moderate adverse events include cutaneous rashes and anaphylactic reactions. If allergic reactions do occur, the use of aPCCs should be suspended, with steroids and antihistamines being administered. The most serious adverse events occurring during treatment with aPCCs are thromboembolic events, especially myocardial infarction; this may occur particularly after the use of high doses of aPCC. Alterations in the results of laboratory tests and/or occasional signs of disseminated intravascular coagulation can appear after administration of these agents. aPCCs should not be used with systemic antifibrinolytics because of the risk of thrombotic complications.
rFVIIa: Non-serious adverse events are rare and those most frequently reported are: pain at the injection site; fever; migraine; vomiting; fluctuations in arterial pressure; and cutaneous reactions of hypersensitivity. Serious treatment-related adverse events tend to be thromboembolic in nature, mainly myocardial or cerebral infarctions, or pulmonary embolisms.
In general, thrombotic complications are rare with both aPCCs and rFVIIa. However, these agents should be used with caution in patients who have a high thrombotic risk 59, 70, 115-117.
Concluding comments
Latin American countries share common economic and social problems. In many of these countries obtaining products for the treatment of hemophilia has not been a priority, resulting in a lack of adequate treatment, and ensuing serious consequences, for patients with hemophilia. However, in recent years, some countries in LA have made huge efforts to improve the care of these patients, developing a national programme and acquiring increasing amounts of therapeutic products to attend to patients' needs. Despite this improvement, the exchange of best practices is not routine within the region and many hemophilia centres seek specialised support from experts from developed countries. The situation is gradually changing as good results from local initiatives begin to appear, and proper support for the treatment of patients with hemophilia can be obtained in neighbouring countries.
One of the objectives of the CLOTTING group is to communicate and encourage the adoption of a good standard of hemophilia care in LA. The purpose of this review is to create awareness of the management of inhibitors in patients with hemophilia. Inhibitors pose serious challenges for physicians, and for patients and their families. Therefore there is an urgent need to know how to properly manage these patients.
The opportunity to share experiences is extremely important in creating the necessary motivation for establishing proper inhibitor treatment. This was attained during the preparation of this document. As a result, the CLOTTING review is based on best current practice and updated published literature as seen from the perspective of Latin American hemophilia specialists.
Conflict of interest: CLOTTING group activities and meetings are supported by an unrestricted educational grant from the Novo Nordisk Latin America Regional Office and affiliates.
Appendix A
Levels of evidence and grading of recommendations based on AHCPR118, 1992.
| Level | Type of evidence |
| Ia | Evidence obtained from meta-analysis of randomised studies |
| Ib | Evidence obtained from at least one randomised controlled trial |
| IIa | Evidence obtained from at least one well-designed controlled study without randomisation |
| IIb | Evidence obtained from at least one other type of well-designed quasi-experimental study |
| III | Evidence obtained from well-designed non-experimental descriptive studies, such as comparative studies, correlation studies and case control studies. |
| IV | Evidence obtained from expert committee reports or opinions and/or clinical experience of respected authorities |
| Grade | Recommendations |
| A (Ia,Ib) | Requires at least one randomised controlled trial as part of the body of literature of overall good quality and consistency addressing the specific recommendation. |
| B (IIa, IIB, III) | Requires availability of well-conducted clinical studies but no randomised clinical trials on the topic of the recommendation. |
| C (IV) | Requires evidence from expert committee reports or opinions and/or clinical experience of respected authorities. Indicates absence of directly applicable studies of good quality. |
Members of the CLOTTING group
1. Dr. Raúl Pérez Bianco*, Instituto de Investigaciones Hematológicas, Academia Nacional de Medicina; Fundación Nacional de Hemofilia, Buenos Aires, Argentina, e-mail:perezbianco@hematologia.anm.edu.ar
2. Dra. Margareth Castro Ozelo* , Hemocentro de Campinas, Universidade Estadual de Campinas, Campinas, Brazil, e-mail: margarethozelo@gmail.com
3. Dr. Paula Ribeiro Villaça, Centro de Hemofilia, Hospital das Clínicas, Universidade de São Paulo, São Paulo, Brazil, e-mail: paulavillaca@yahoo.com
4. Dra. Maria Helena Solano, Hospital de San Jose, Fundación Universitaria Ciencias de la Salud, Bogotá, Colombia, e-mail: mhsolano@coldecon.net.co
5. Dr. Guillermo Jiménez Cruz, Hospital México, Caja Costaricense de Seguro Social, San José, Costa Rica, e-mail: jimeno1951@yahoo.com.mx
6. Dr. Carlos Martínez Murillo, Unidad Médica de Alta Especialidad, Centro Médico Nacional Siglo XXI, Ciudad de México, México, e-mail: carlmarz@prodigy.net.mx
7. Dr. Jaime García Chávez, Unidad Médica de Alta Especialidad, Centro Médico Nacional La Raza, Ciudad de México, México, e-mail: jaimegch@prodigy.net.mx
8. Dr. Saúl Mendoza, Hospital Nacional Edgardo Rebagliati Martins - EsSalud Lima, Perú, e-mail: smendoza@clathtperu.org
9. Dr. Ismael Rodríguez Grecco, Servicio de Medicina Transfusional, Centro Hospitalario Pereira Rossell, Montevideo, Uruguay, e-mail: isro@adinet.com.uy
10. Dr. Arlette Ruiz-Sáez*, Centro Nacional de Hemofilia, Banco Metropolitano de Sangre, Caracas, Venezuela, e-mail: arletteruizsaez@hotmail.com
*Committee member from a centre with the capability to perform genetic testing.
1. Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992; 339: 594-8. [ Links ]
2. Katz J. Prevalence of factor IX inhibitors among patients with haemophilia B: results of a large-scale North American study. Haemophilia 1996; 2: 28-31. [ Links ]
3. Goudemand J. Pharmaco-economic aspects of inhibitor treatment. Eur J Haematol 1998; 61: 24-7. [ Links ]
4. World Hemophilia Federation Report on Global Survey 2006. World Federation of Hemophilia, 2007. On www.wfh.org; consulted on 23/04/2008. [ Links ]
5. Rieger A, Roisenberg I. Prevalence of factor VIII inhibitors in patients with hemophilia A in Brazil. Thromb Haemost 1999; 81: 475-6. [ Links ]
6. Fontes EM, Amorim L, Carvalho SM, Farah MB. Hemophilia care in the state of Rio de Janeiro, Brazil. Rev Panam Salud Pública 2003; 13: 124-8. [ Links ]
7. Izquierdo-Ramírez J, Contreras-Mulato EL, Sotelo-Ham EI et al. Incidence of inhibitors in children with hemophilia A . Bol Med Hosp Infant Méx 1988; 45: 578-82. [ Links ]
8. Boadas A, Ruiz-Sáez A, Arguello A, de Bosch N. Prevalence and acute bleeding treatment of allo and auto FVIII and FIX antibodies cases in Venezuela. Haemophilia 2004; 10: 56. [ Links ]
9. Wight J, Paisley S. The epidemiology of inhibitors in hemophilia A: a systematic review. Haemophilia 2003; 9: 418-35. [ Links ]
10. Delivery of treatment for hemophilia. Report of a joint WHO/WFH/ISTH meeting. World Health Organization, 2002. [ Links ]
11. Hay CR, Brown S, Collins PW, Keeling DM, Liesner R. The diagnosis and management of factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre Doctors Organization. Br J Haematol 2006; 133: 591-605. [ Links ]
12. Ewing NP, Kasper CK. In vitro detection of mild inhibitors to factor VIII in hemophilia. Am J Clin Pathol 1982; 77: 749-52. [ Links ]
13. Verbruggen B, Novakova I, Wessels H, Boezeman J, van den Berg M, Mauser-Bunschoten E. The Nijmegen modification of the Bethesda assay for factor VIII:C inhibitors: improved specificity and reliability. Thromb Haemost 1995; 73: 247-51. [ Links ]
14. Giles AR, Verbruggen B, Rivard GE, Teitel J, Walker I. A detailed comparison of the performance of the standard versus the Nijmegen modification of the Bethesda assay in detecting factor VIII:C inhibitors in the hemophilia A population of Canada. Association of Hemophilia Centre Directors of Canada. Factor VIII/IX Subcommittee of Scientific and Standardization Committee of International Society on Thrombosis and Haemostasis. Thromb Haemost 1998; 79: 872-5. [ Links ]
15. Verbruggen B, van Heerde W, Novakova I, Lillicrap D, Giles A. A 4% solution of bovine serum albumin may be used in place of factor VIII:C deficient plasma in the control sample in the Nijmegen modification of the Bethesda factor VIII:C inhibitor assay. Thromb Haemost 2002; 88: 362-4. [ Links ]
16. White GC, II, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost 2001; 85: 560. [ Links ]
17. Kitchen S, McCraw A. Diagnosis of hemophilia and other bleeding disorders. A laboratory manual. World Federation of Hemophilia, 2000. [ Links ]
18. Available at: http://www.med.unc.edu/isth/. Accessed November 2005. [ Links ]
19. Available at: http://www.wfh.org/. Accessed November 2005. [ Links ]
20. Oldenburg J, Brackmann HH, Schwaab R. Risk factors for inhibitor development in hemophilia A. Haematologica 2000; 85: 7-13. [ Links ]
21. Rossetti LC, Candela M, Pérez Bianco R, de Tezanos Pinto M, Western A, Goodeve A et al. Analysis of factor VIII gene intron 1 inversion in Argentinean families with severe hemophilia A and a review of the literature. Blood Coagul Fibrinolysis 2004;15: 569-72. [ Links ]
22. Santos A, Montalva O, Thomas S, Veiga M, De Paula E, Ozelo M. Genetic and ethnic aspects related to the development of inhibitors among Brazilian patients with hemophilia from five distinct geographical regions in Brazil. Haemophilia 2006; 12: 1-154. [ Links ]
23. Mantilla-Capacho JM, Beltrán-Miranda CP, Luna-Záizar H, et al. Frequency of intron 1 and 22 inversions of Factor VIII gene in Mexican patients with severe Hemophilia A. Am J Hematol 2007; 82: 283-7. [ Links ]
24. Guidelines for the management of hemophilia. World Federation of Hemophilia, 2005. [ Links ]
25. Girolami A, Luzzatto G, Varvarikis C, Pellati D, Sartori R, Girolami B. Main clinical manifestations of a bleeding diathesis: an often disregarded aspect of medical and surgical history taking. Haemophilia 2005; 11: 193-202. [ Links ]
26. Inhibitor Subcommittee of the Association of Hemophilia Clinic Directors of Canada. Suggestions for the management of FVIII inhibitors, revised edition. In: Treatment of Hemophilia Monograph. World Federation of Hemophilia, 2000. [ Links ]
27. Kasper C. Diagnosis and management of inhibitors to factors VIII and IX. In: Treatment of Hemophilia Monograph. World Federation of Hemophilia, 2004. [ Links ]
28. Gringeri A, Mannucci PM; Italian Association of Haemophilia Centres. Italian guidelines for the diagnosis and treatment of patients with hemophilia and inhibitors. Haemophilia 2005; 11: 611-9. [ Links ]
29. Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (NovoSeven) in haemophiliacs with inhibitors. Thromb Haemost 1998; 80: 912-8. [ Links ]
30. Ingerslev J, Sneppen O, Hvid I, Fredberg U, Kristensen HL, Sindet-Petersen S. Treatment of acute bleeding episodes with rFVIIa. Vox Sang 1999; 77: 42-6. [ Links ]
31. Santagostino E, Gringeri A, Mannucci PM. Home treatment with recombinant activated factor VII in patients with factor VIII inhibitors: the advantages of early intervention. Br J Haematol 1999; 104: 22-6. [ Links ]
32. Negrier C, Goudemand J, Sultan Y, Bertrand M, Rothschild C, Lauroua P. Multicenter retrospective study on the utilization of FEIBA in France in patients with factor VIII and factor IX inhibitors. French FEIBA Study Group. Factor Eight Bypassing Activity. Thromb Haemost 1997; 77: 1113-9. [ Links ]
33. Astermark J, Donfield SM, DiMichelle DM, et al. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor - The Feiba© NovoSeven® Comparative Study (FENOC). Blood 2007; 109: 546-51. [ Links ]
34. Young G, Shafer FE, Rojas P, Seremetis S. Single 270 µg kg-1 dose rFVIIa vs. standard 90 µg kg-1 dose rFVIIa and APCC for home treatment of joint bleeds in hemophilia patients with inhibitors: a randomized comparison. Haemophilia 2008, 14: 287-94. [ Links ]
35. Ozelo MC, Villaça PR, De Almeida JO, et al. A cost evaluation of treatment alternatives for mild-to-moderate bleeding episodes in patients with hemophilia and inhibitors in Brazil. Haemophilia 2007;13: 462-9. [ Links ]
36. Kenet G, Lubetsky A, Luboshitz J, Martinowitz U. A new approach to treatment of bleeding episodes in young hemophilia patients: a single bolus megadose of recombinant activated factor VII (NovoSeven). J Thromb Haemost 2003; 1: 450-5. [ Links ]
37. Parameswaran R, Shapiro AD, Gill JC, Kessler CM. Dose effect and efficacy of rFVIIa in the treatment of hemophilia patients with inhibitors: analysis from the Hemophilia and Thrombosis Research Society Registry. Haemophilia 2005; 11: 100-6. [ Links ]
38. Kavakli K, Makris M, Zulfikar B, Erhardtsen E, Abrams ZS, Kenet G. Home treatment of haemarthroses using single dose regimen of recombinant activated factor VII in patients with hemophilia and inhibitors. A multi-centre, randomized, double blind, cross-over trial. Thromb Haemost 2006; 95: 600-5. [ Links ]
39. Santagostino E, Mancuso ME, Rocino A, Mancuso G, Scaraggi F, Mannucci PM. A prospective randomized trial of high and standard dosages of recombinant factor VIIa for treatment of haemarthroses in hemophiliacs with inhibitors. J Thromb Haemost 2006; 4: 367-71 [ Links ]
40. Schneiderman J, Nugent DJ, Young G. Sequential therapy with activated prothrombin complex concentrate and recombinant factor VIIa in patients with severe hemophilia and inhibitors. Haemophilia 2004; 10: 347-51. [ Links ]
41. Lusher JM, Shapiro SS, Palascak JE, Rao AV, Levine PH, Blatt PM. Efficacy of prothrombin-complex concentrates in hemophiliacs with antibodies to factor VIII: a multicenter therapeutic trial. NEJM 1980; 303: 421-5. [ Links ]
42. Teitel J, Berntorp E, Collins P, et al. A systematic approach to controlling problem bleeds in patients with severe congenital hemophilia A and high-titre inhibitors. Haemophilia 2007;13: 256-63. [ Links ]
43. Lloyd Jones M, Wight J, Paisley S, Knight C. Control of bleeding in patients with hemophilia A with inhibitors: a systematic review. Haemophilia 2003; 9: 464-520. [ Links ]
44. Ewenstein BM, Takemoto C, Warrier I, et al. Nephrotic syndrome as a complication of immune tolerance in hemophilia B. Blood 1997; 89: 1115-6. [ Links ]
45. Warrier I, Lenk H, Saidi P, Pollmann H, Tengborn L, Berntorp E. Nephrotic syndrome in hemophilia B patients with inhibitors. Haemophilia 1998; 4: 248. [ Links ]
46. Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. Haemophilia 1998; 4: 574-6. [ Links ]
47. Strawczynski H, Stachewitsch A, Morgenstern G, Shaw ME. Delivery of care to hemophilic children: home care versus hospitalization. Pediatrics 1973; 51: 986-91. [ Links ]
48. Rabiner SF, Telfer MC, Fajardo R. Home transfusions of hemophiliacs. JAMA 1972; 221: 885-7. [ Links ]
49. Levine P. The home therapy program at the New England area hemophilia center. Scand J Haematol 1977; 31: 37-51. [ Links ]
50. Soucie JM, Symons J, IV, Evatt B, Brettler D, Huszti H, Linden J. Home-based factor infusion therapy and hospitalization for bleeding complications among males with hemophilia. Haemophilia 2001; 7: 198-206. [ Links ]
51. Solovieva S. Clinical severity of disease, functional disability and health-related quality of life. Three-year follow-up study of 150 Finnish patients with coagulation disorders. Haemophilia 2001; 7: 53-63. [ Links ]
52. Teitel JM, Barnard D, Israels S, Lillicrap D, Poon MC, Sek J. Home management of hemophilia. Haemophilia 2004; 10: 118-33. [ Links ]
53. Ingerslev J, Thykjær H, Scheibel E. Approaches towards successful home treatment in patients with inhibitors. Eur J Haematol 1998: 61: 11-4. [ Links ]
54. Young G, McDaniel M, Nugent DJ. Prophylactic recombinant factor VIIa in hemophilia patients with inhibitors. Haemophilia 2005; 11: 203-7. [ Links ]
55. Saxon BR, Shanks D, Jory CB, Williams V. Effective prophylaxis with daily recombinant factor VIIa (rFVIIa-Novoseven) in a child with high titre inhibitors and a target joint. Thromb Haemost 2001; 86: 1126-7. [ Links ]
56. Konkle BA, Ebbesen LS, Erhardtsen E, et al. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost 2007; 5: 1904-13. [ Links ]
57. Hoots WK, Ebbesen LS, Konkle BA, et al. Secondary prophylaxis with recombinant activated factor VII improves health-related quality of life of hemophilia patients with inhibitors. Haemophilia 2008; 14: 466-75. [ Links ]
58. Leissinger CA. Prevention of bleeds in hemophilia patients with inhibitors: emerging data and clinical direction. Am J Hematol 2004; 77: 187-93. [ Links ]
59. Luu H, Ewenstein B. FEIBA safety profile in multiple modes of clinical and home-therapy application. Haemophilia 2004; 10: 10-6. [ Links ]
60. Leissinger CA, Becton DL, Ewing NP, Valentino LA. Prophylactic treatment with activated prothrombin complex concentrate (FEIBA) reduces the frequency of bleeding episodes in paediatric patients with hemophilia A and inhibitors. Haemophilia 2007; 13: 249-55. [ Links ]
61. Villar A, Aronis S, Morfini M, et al. Pharmacokinetics of activated recombinant coagulation factor VII (NovoSeven©) in children vs. adults with hemophilia A. Haemophilia 2004; 10: 352-9 [ Links ]
62. Klitgaard T, Nielsen TG. Overview of the human pharmacokinetics of recombinant activated factor VII. Br J Clin Pharmacol 2008; 65: 3-11. [ Links ]
63. Rodriguez-Merchan EC, Rocino A, Ewenstein B, et al. Consensus perspectives on surgery in hemophilia patients with inhibitors: summary statement. Haemophilia 2004; 10: 50-2. [ Links ]
64. Hilgartner MW. Factor replacement therapy. In: Hilgartner MW, Pochedly C (eds): Hemophilia in the Child and Adults. New York, Raven Press Ltd., 1989, pp 1-26. [ Links ]
65. Morris PJ, Wood WC (eds.): Oxford Textbook of Surgery, 2nd edition. Oxford, Oxford University Press, 2000. [ Links ]
66. Rickard KA. Guidelines for therapy and optimal dosages of coagulation factors for treatment of bleeding and surgery in hemophilia. Haemophilia 1995; 1: 8-13. [ Links ]
67. Shapiro A, Gilchrist GS, Hoots WK, Cooper HA, Gastineau DA. Prospective, randomised trial of two doses of rFVIIa (NovoSeven) in hemophilia patients with inhibitors undergoing surgery. Thromb Haemost 1998; 80: 773-8. [ Links ]
68. Hvid I, Rodriguez-Merchan EC. Orthopaedic surgery in haemophilic patients with inhibitors: an overview. Haemophilia 2002; 8: 288-91. [ Links ]
69. Rodriguez-Merchan EC, Rocino A. Literature review of surgery management in inhibitor patients. Haemophilia 2004; 10: 22-9. [ Links ]
70. Abshire T, Kenet G. Recombinant factor VIIa: review of efficacy, dosing regimens and safety in patients with congenital and acquired factor VIII or IX inhibitors. J Thromb Haemost 2004; 2: 899-909. [ Links ]
71. Ingerslev J. Efficacy and safety of Recombinant Factor VIIa in the prophylaxis of bleeding in various surgical procedures in hemophilic patients with factor VIII and factor IX inhibitors. Semin Thromb Hemost 2000; 26: 425-32. [ Links ]
72. Tjønnfjord GE, Brinch L, Gedde-Dahl T, III, Brosstad FR. Activated prothrombin complex concentrate (FEIBA) treatment during surgery in patients with inhibitors to FVIII/IX. Haemophilia 2004; 10: 174-8. [ Links ]
73. Ingerslev J, Sorensen B. Role of recombinant activated factor VII as hemostatic support in orthopedic surgery. TATM 2006; 8: 35-42. [ Links ]
74. Obergfell A, Auvinen MK, Mathew P. Recombinant activated factor VII for haemophilia patients with inhibitors undergoing orthopaedic surgery: a review of the literature. Haemophilia 2008; 14: 233-41. [ Links ]
75. Ewenstein BM, Valentino LA, Journeycake JM, et al. Consensus recommendations for use of central venous access devices in hemophilia. Haemophilia 2004; 10: 629-48. [ Links ]
76. Morado M, Jimenez-Yuste V, Villar A, et al. Complications of central venous catheters in patients with hemophilia and inhibitors. Haemophilia 2001; 7: 551-6. [ Links ]
77. Bollard CM, Teague LR, Berry EW, Ockelford PA. The use of central venous catheters (portacaths) in children with hemophilia. Haemophilia 2000; 6: 66-70. [ Links ]
78. O'Connell N, Mc Mahon C, Smith J, et al. Recombinant factor VIIa in the management of surgery and acute bleeding episodes in children with hemophilia and high responding inhibitors. Br J Haematol 2002; 116: 632-5. [ Links ]
79. Cooper HA, Jones CP, Campion E, Roberts HR, Hedner U. Rationale for the use of high dose rFVIIa in a high-titre inhibitor patient with hemophilia B during major orthopaedic procedures. Haemophilia 2001; 7: 517-22. [ Links ]
80. Colowick AB, Bohn RL, Avorn J, Ewenstein BM. Immune tolerance induction in hemophilia patients with inhibitors: costly can be cheaper. Blood 2000; 96: 1698-702. [ Links ]
81. Brackmann HH, Gormsen J. Massive factor-VIII infusion in haemophiliac with factor-VIII inhibitor, high responder. Lancet 1977; 2: 933. [ Links ]
82. Key NS. Inhibitors in congenital coagulation disorders. Br J Haematol 2004; 127: 379-91. [ Links ]
83. Nilsson IM, Berntorp E, Zettervall O. Induction of immune tolerance in patients with hemophilia and antibodies to factor VIII by combined treatment with intravenous IgG, cyclophosphamide, and factor VIII. NEJM 1988; 318: 947-50. [ Links ]
84. Mathias M, Khair K, Hann I, Liesner R. Rituximab in the treatment of alloimmune factor VIII and IX antibodies in two children with severe hemophilia. Br J Haematol 2004; 125: 366-8. [ Links ]
85. Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004; 103: 4424-8. [ Links ]
86. Carcao M, Ungar WJ, Feldman BM. Cost-utility analysis in evaluating prophylaxis in hemophilia. Haemophilia 2004; 10: 50-7. [ Links ]
87. Curtin J, Misra A, Teo J, Webster B, Lammi A. Use of Rituximab as an alternative strategy for the management of difficult high titre inhibitors in children with hemophilia A. Haemophilia 2004; 10: 57. [ Links ]
88. DiMichele D. Immune tolerance therapy dose as an outcome predictor. Haemophilia 2003; 9: 382-6. [ Links ]
89. Mariani G, Kroner B. Immune tolerance in hemophilia with factor VIII inhibitors: predictors of success. Haematologica 2001; 86: 1186-93. [ Links ]
90. DiMichele D. Inhibitors: resolving diagnostic and therapeutic dilemmas. Haemophilia 2002; 8: 280-7. [ Links ]
91. Lenk H. The German Registry of immune tolerance treatment in hemophilia-1999 update. Haematologica 2000; 85: 45-7. [ Links ]
92. Haya S, Lopez MF, Aznar JA, Batlle J. Immune tolerance treatment in hemophilia patients with inhibitors: the Spanish Registry. Haemophilia 2001; 7:154-9. [ Links ]
93. DiMichele DM, Hoots WK, Pipe SW, Rivard GE, Santagostino E. International workshop on immune tolerance induction: consensus recomendations. Haemophilia 2007; 13: 1-22. [ Links ]
94. Kreuz W, Mentzer D, Auerswald G, Becker S, Joseph-Steiner J. Successful immune tolerance therapy of FVIII inhibitor in children after changing from high to intermediate purity FVIII concentrate. Haemophilia 1996; 2: 19. [ Links ]
95. Rocino A, Papa ML, Salerno E, Capasso F, Miraglia E, de Biasi R. Immune tolerance induction in hemophilia A patients with high-responding inhibitors to factor VIII: experience at a single institution. Haemophilia 2001; 7: 33-8. [ Links ]
96. DiMichele D, Rivard G, Hay C, Antunes S. Inhibitors in hemophilia: clinical aspects. Haemophilia 2004; 10: 140-5. [ Links ]
97. Mauser-Bunschoten EP, Nieuwenhuis HK, Roosendaal G, van den Berg HM. Low-dose immune tolerance induction in hemophilia A patients with inhibitors. Blood 1995; 86: 983-8. [ Links ]
98. Almeida J, Paula JC, Toscano R. Immune tolerance such as salvage therapy in severe hemophilia A patient with ultra high-responders inhibitors. Haemophilia 2002; 8: 538. [ Links ]
99. Solano MH, Ramírez C, Parra L. Tratamiento de inhibidores del factor VIII en hemofilia. Acta Med Colomb 1998; 23: 193. [ Links ]
100. Carneiro JDA, Bassit RP, Villaça PR, Sandoval EPN, Silva CSSS, D'amico EA. Low-dose immune tolerance induction in hemophilia A children with inhibitors. Haemophilia 2002; 8: 538-9. [ Links ]
101. Wight J. Paisley S, Knight C. Immune tolerance induction in patients with hemophilia A with inhibitors: a systematic review. Haemophilia 2003; 9: 436-63. [ Links ]
102. Tengborg L, Hansson S, Fasth A, Lübeck PO, Berg A, Ljung R. Anaphylactoid reactions and nephrotic syndrome -a considerable risk during factor IX treatment in patients with hemophilia B and inhibitors: a report on the outcome in two brothers. Haemophilia 1998; 4: 854-9. [ Links ]
103. Schulman S. Safety, efficacy and lessons from continuous infusion with rFVIIa. Haemophilia 1998; 4: 564-7. [ Links ]
104. Schulman S. Continuous infusion of recombinant factor VIIa in hemophilic patients with inhibitors: safety, monitoring, and cost effectiveness. Semin Thromb Hemost 2000; 26: 421-4. [ Links ]
105. Pruthi RK, Mathew P, Valentino LA, Sumner MJ, Seremetis S, Hoots WK. Haemostatic efficacy and safety of bolus and continuous infusion of recombinant factor VIIa are comparable in hemophilia patients with inhibitors undergoing major surgery. Results from an open-label, randomized, multicenter trial. Thromb Haemost. 2007; 98: 726-32. [ Links ]
106. Smith MP, Ludlam CA, Collins PW, et al. Elective surgery on factor VIII inhibitor patients using continuous infusion of recombinant activated factor VII: plasma factor VII activity of 10 IU/ml is associated with an increased incidence of bleeding. Thromb Haemost 2001; 86: 949-53. [ Links ]
107. Santagostino E, Morfini M, Rocino A, Baudo F, Scaraggi FA, Gringeri A. Relationship between factor VII activity and clinical efficacy of recombinant factor VIIa given by continuous infusion to patients with factor VIII inhibitors. Thromb Haemost 2001; 86: 954-8. [ Links ]
108. Mauser-Bunschoten EP, Koopman MM, Goede-Bolder AD, et al. Efficacy of recombinant factor VIIa administered by continuous infusion to hemophilia patients with inhibitors. Haemophilia 2002; 8: 649-56. [ Links ]
109. Ludlam CA, Smith MP, Morfini M, Gringeri A, Santagostino E, Savidge GF. A prospective study of recombinant activated factor VII administered by continuous infusion to inhibitor patients undergoing elective major orthopaedic surgery: a pharmacokinetic and efficacy evaluation. Br J Haematol 2003; 120: 808-13. [ Links ]
110. Escobar MA. Recombinant Factor VIIa: the possibilities for monitoring. TATM 2003; 5: 51-4. [ Links ]
111. Young G, Ebbesen LS, Viuff D, et al. Evaluation of thromboelastography for monitoring recombinant activated factor VII ex vivo in hemophilia A and B patients with inhibitors: a multicentre trial. Blood Coagul Fibrinolysis 2008; 19: 276-82. [ Links ]
112. Lak M, Scharling B, Blemings A, et al. Evaluation of rFVIIa (NovoSeven) in Glanzmann patients with thromboelastogram. Haemophilia 2008;14:103-10. [ Links ]
113. Sørensen B, Ingerslev J. Whole blood clot formation phenotypes in hemophilia A and rare coagulation disorders. Patterns of response to recombinant factor VIIa. J Thromb Haemost 2004; 2: 102-110. [ Links ]
114. Varadi K, Turecek PL, Schwarz HP. Thrombin generation assay and other universal tests for monitoring hemophilia therapy. Haemophilia 2004; 10: 17-21. [ Links ]
115. Aledort LM. Comparative thrombotic event incidence after infusion of recombinant factor VIIa versus factor VIII inhibitor bypass activity. J Thromb Haemost 2004; 2: 1700-8. [ Links ]
116. Sallah S, Isaksen M, Seremetis S, Payne Rojkjaer L. Comparative thrombotic event incidence after infusion of recombinant factor VIIa vs. factor VIII inhibitor bypass activity: a rebuttal. J Thromb Haemost 2005; 3: 820-2. [ Links ]
117. Roberts HR, Monroe DM, III, Hoffman M. Safety profile of recombinant factor VIIa. Semin Hematol 2004; 41: 101-8. [ Links ]
118. Agency of Health Care Policy and Research. Acute pain management: operative or medical procedures and trauma (Agency of Health Care Policy and Research publications). Clinical Pharmacology 1992; 11: 391-414. [ Links ]
Received: 14-05-2008
Accepted: 04-06-2008

