










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680On-line version ISSN 1669-9106
Medicina (B. Aires) vol.68 no.5 Ciudad Autónoma de Buenos Aires Sept./Oct. 2008
Poliquistosis renal: todo ocurrió por un pelito
En 1898, Zimmerman, en la época de los trabajos de un único autor, llamó la atención sobre algo que sus epígonos consolidaron: que todas las células, desde el alga verde Chlamydomonas y el gusano Caenorhabditis elegans hasta las células humanas -y especialmente las renales- poseían un cilium no móvil, que emergía enhiesto y solitario, en búsqueda de alguna función desconocida (Fig. 1). Dado que debieron transcurrir 63 años para verificar ese hallazgo por microscopía electrónica, Wheatly se pregunta recientemente, entre dudoso e irreverente, si Zimmerman no tuvo el mismo accidente que él con el microscopio convencional muchos años después: el de una burbuja del tamaño adecuado en el lugar adecuado del preparado histológico, que agregó algunas dioptrías extras para reconocer el cilium1.
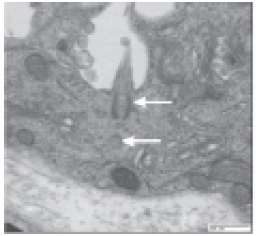
Fig 1. Microscopía electrónica de una neurona sensorial con su cilium (flecha superior), por encima del cuerpo basal (flecha inferior). (Cortesía de N. Katsanis, John Hopkins University)
Los primeros investigadores en poliquistosis renal, si bien pudieron haber tenido noticias del cilium, no pensaron en buscar conexión alguna entre éste y la enfermedad. Quizá al principio primó un enfoque molecular, con la esperanza de que el descubrimiento del gen llevaría a la identificación de la proteína involucrada, de ahí a la función deteriorada y -cui bono- al tratamiento de la enfermedad. Este era un enfoque evolucionado, muy distinto al de buscar y encontrar la proteína ignorando al gen; empresa esta sí laboriosa, tanto que llevó a Leloir a acuñar el axioma general "que en los líquidos biológicos había muchas sustancias sin nombre y muchos nombres sin sustancia". Este enfoque del gen a la proteína llevó entonces, en 1994 y 1996, al descubrimiento de dos genes, PDK1 y PKD2, con sus correspondientes proteínas deducidas, las policistinas 1 y 22,3.
Paralelamente a esta línea molecular, otros investigadores se hicieron otra pregunta: ¿por qué un quiste, que es al fin un "pedacito" del túbulo renal, se vuelve una entidad autónoma, deja de absorber líquido como un nefrón sabe hacerlo y pasa a secretarlo grotescamente, en una suerte de reptilización, atendiendo a la importancia del proceso secretorio en estos animales?4. Esta línea, que ciertamente no desconocía el cilium ni los genes descriptos, concibió un modelo celular, el de cotransportadores de cloro/sodio/potasio en la membrana basal y canales de cloro en la membrana luminal (Fig. 2), que se ayudan mutuamente para crear un flujo unidireccional de fluido hacia la luz del quiste y hacerlo crecer así en forma exponencial5. Puede verse así en el panel derecho de la Fig. 2, que el ion cloro entra a la célula a través del cotransportador NKCC1 y sale de la célula a través de canales CFTR (cystic fibrosis transmembrane conductance regulator) y purinérgicos. Este transporte crea una diferencia de potencial eléctrico -lumen negativo- que atrae al ion sodio por la vía paracelular y luego al agua. Este "mundo del revés" -compárese con el panel de la izquierda- hace crecer al quiste, proceso que se manifiesta en forma precisa al medir el volumen renal por imágenes, herramienta que en la práctica clínica se ha convertido en un marcador de progresión en los largos años en que el clearance de creatinina se mantiene normal. Este enfoque tubular del problema llevó a otra sorpresa, típica de la fisiología sistémica, la de que las células del quiste mostraban un contenido de AMPc mayor que el de las células tubulares no quísticas6. Y esto fue importante, ya que el AMPc es estimulado por la presencia de vasopresina, cuyo receptor V2 está localizado en la membrana basolateral (Fig. 2), abriendo así el camino para el uso de inhibidores competitivos del receptor de vasopresina -como el OPC-31260 y la somatostatina- que han comenzado a usarse recientemente en ensayos clínicos de fase III7,8. Todo este proceso de secreción desenfrenada va acompañado también de proliferación epitelial, que ayuda al quiste a crecer, mecanismo en parte dependiente de la vía celular mTOR, proteína blanco de la rapamicina, también ésta en uso en ensayos clínicos humanos9.
Fig. 2.- Mecanismos de transporte electrolítico en las células principales del túbulo colector cortical. A la izquierda el fenotipo normal. ENaC, canal epitelial de sodio; ROMK, canal de potasio de la medular externa; V2R, receptor tipo 2 de la vasopresina; VP, vasopresina; AC, adenilato ciclasa. A la derecha el fenotipo del quiste. CFTR, canal regulador de conductancia (al cloro) de la fibrosis quística; NKCC2, cotransportador 2Cl-Na+-K+. Adaptado de Grantham5.
Volvamos ahora al cilium; no se conocía su función en el año 1994, año del descubrimiento del gen PKD1. Pero en ese año aparece la primera evidencia experimental de que el cilium tiene algo que ver con la enfermedad y que tiene una función: el ratón Tg737orpk , con una mutación en el gen Tg737, tiene como resultado una poliquistosis renal10. Luego viene una avalancha de eventos, que a través del gen IFT88 de la Chlamydomonas lleva a asegurar que el defecto primario de aquel ratón era una incapacidad para armar las cilias primarias, que el nematodo Caenorhabditis elegans posee homólogos de la policistina 1 y 2 en sus cilias inmóviles y que ambas policistinas se encuentran alojadas en las cilias renales. ¿Qué hace el cilium de las células renales tubulares normales? Trafica información desde el centriolo hasta su punta y esa punta actúa como un mecanosensor, ya que al ser "doblada" por el fluido tubular, envía señales que regulan los pooles de calcio y AMPc intracelulares y mantienen quiescentes a sus células. Imaginemos ese escenario con un cilium mutado en algunas de sus estructuras y entenderemos por qué esto puede llevar a la desdiferenciación, proliferación y apoptosis características de la poliquistosis renal. En otras palabras, aunque el cilium parece morfológicamente normal en esta enfermedad, falla en su capacidad de sensar estímulos mecánicos y químicos. A modo de cascada, otras cuatro enfermedades quísticas, que tienen proteínas mutadas en el cilium, han entrado a engrosar el cuadro de las ciliopatías, como la forma recesiva de la poliquistosis renal y las nefronoptisis, todas a la espera de clarificar sus mecanismos celulares y de un pronóstico mejor. Así y todo, y como es obvio en biología, no todas las enfermedades quísticas tienen necesariamente un defecto en las proteínas ciliares; recientemente, en un estudio cooperativo en una familia argentina con poliquistosis hepática autonómica dominante -y sin poliquistosis renal- comprobamos que la proteína mutada estaba localizada en un componente de la maquinaria de translocación en el retículo endoplásmico, lo que sugiere también la importancia de proteínas no ciliares en la formación de quistes11.
La poliquistosis renal autosómica dominante es la enfermedad renal genética más frecuente, constituye aproximadamente 7% de los cerca de 25.000 pacientes en diálisis crónica en Argentina12, disminuye la calidad de vida de numerosas familias y su tratamiento requiere alrededor de 80 millones de pesos anuales en nuestro país. Es de desear que, si bien será difícil atacar "de cuajo" un problema biológico tan fundamental, se pueda disminuir la velocidad de progresión de la enfermedad a ritmos que no requieran tratamientos sustitutivos renales.
Entretanto, y como ocurre muchas veces en ciencia, se necesitaron unos pocos 100 años para que distintas líneas de investigación se entrecruzaran y se reconocieran como partes de un nuevo orden. Ya lo dijo J. Bronowski con belleza en uno de sus ensayos, ..."toda la ciencia es la búsqueda de la unidad en las semejanzas escondidas".
Rodolfo S. Martin1, Pablo J. Azurmendi2
1Facultad de Ciencias Biomédicas, Universidad Austral
2Instituto de Investigaciones Médicas Alfredo Lanari, Universidad de Buenos Aires
e-mail: rsmartin@mail.retina.ar
1. Wheatley DN. Landmarks in the first hundred years of primary (9+0) cilium research. Cell Biol Int 2005; 29: 333-9. [ Links ]
2. The European Polycystic Kidney Disease Consortium. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region of chromosome 16. Cell 1994; 77: 881-94. [ Links ]
3. Mochizuki T, Wu G, Hayashi T et al. PKD2, a gen for polycystic kidney disease that encodes an integral membrane protein. Science 1996; 272:1339-42. [ Links ]
4. Grantham JJ, Wallace DP. Return of the secretory kidney. Am J Physiol 2002; 282: F1-F9. [ Links ]
5. Grantham JJ. Lillian Jean Kaplan International Prize for advancement in the understanding of polycystic kidney disease. Understanding polycystic kidney disease: a systems biology approach. Kidney Int 2003; 64:1157-62. [ Links ]
6. Belibi FA, Reif G, Wallace DP, et al. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int 2004; 66: 964-73. [ Links ]
7. Gattone VH2, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 1323-26. [ Links ]
8. Rugenetti P, Remuzzi A, Ondei P, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal polycystic kidney disease. Kidney Int 2005; 206-16. [ Links ]
9. Tao Y, Kim J, Schrier RW, Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol 2005; 16: 46-51. [ Links ]
10. Moyer JH, Lee-Tischler MJ, Kwon H-Y, et al. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 1994; 264:1329-33. [ Links ]
11. Davila S, Furu L, Gharavi AG, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet 2004; 36: 575-7. [ Links ]
12. Instituto Nacional Central Unico Coordinador de Ablación e Implante (INCUCAI). Sistema Nacional de Información de Procuración y Transplante de la R. Argentina. En: http://sintra.incucai.gov.ar; Consultado el 4/8/08. [ Links ]

