










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) v.69 n.1 supl.1 Ciudad Autónoma de Buenos Aires 2009
Miastenia gravis juvenil
Oscar Papazian, Israel Alfonso, Nayle Araguez
Departamento de Neurología, Miami Children's Hospital, Miami FL, USA
Dirección postal: Dr. O. Papazian, Departamento de Neurología, Miami Children's Hospital, Miami, Florida, USA. e-mail: Oscar.Papazian@mch.com
Resumen
La miastenia gravis juvenil (MGJ) es un trastorno crónico auto inmune en el cual existen anticuerpos séricos que al unirse a los receptores de acetilcolin nicotínicos de la membrana muscular de la placa motora alteran la transmisión neuromuscular. El resultado es fatiga muscular precoz con progresión a la parálisis durante estados de contracción muscular iterativos (movimientos) o sostenidos (posturas) y más raramente parálisis permanente durante el reposo. Los músculos inervados por los nervios craneales, especialmente los extraoculares y elevadores de los párpados, tienen más tendencia a la debilidad muscular persistente que los inervados por otros pares craneales y las extremidades. Las formas clínicas de presentación son generalizadas, oculares y respiratorias. El diagnóstico se sospecha mediante la anamnesia, la fatiga anormal se comprueba mediante el examen físico y la estimulación eléctrica iterativa del nervio que inerva al músculo afectado pero no paralizado. Se corrobora mediante la administración de inhibidores de la acetilcolin esterasa (IACE) que al aumentar la cantidad de acetilcolin en la hendidura sináptica, corrigen la fatiga o la debilidad muscular transitoriamente. Se hace el diagnóstico de certeza mediante la demostración sérica de anticuerpos contra los receptores de acetilcolin (ACRA). El tratamiento es a largo plazo sintomático con IACE y etiopatogénico con inmunosupresores, plasmaféresis, gamma globulina endovenosa y timectomía. El curso es crónico. La remisión espontánea o después de tratamiento sintomático o etiopatogénico ocurre entre 1-10 años respectivamente. La mortalidad es prácticamente nula aun durantes las crisis miastenias gracias a la educación de padres, pacientes y público en general sobre el tema, al desarrollo del sistema de respuesta rápida de auxilio domiciliario y las unidades de cuidados intensivos y el empleo de la ventilación asistida profiláctica, plasmaféresis y administración endovenosa de gamma globulina.
Palabras claves: Miastenia gravis; Diagnóstico; Tratamiento; ACRA
Abstract
Juvenile myasthenia gravis. Juvenile myasthenia gravis is a chronic autoimmune disorder which occurs when serum antibodies combine with nicotinic acetylcholine receptors at the muscle membrane of the motor endplate imparing the neuromuscular transmission. It results in early muscle fatigability with progression to a complete paralysis during repetitive muscle contraction (movements) or steady muscle contraction (postures), and less common persistent paralysis at rest. The cranial nerves, mainly the one innervating the extraoccular and palpebral levator, are the most susceptible to permanent weakness and paralysis at rest. Initial clinical presentations are generalized, ocular and respiratory forms. The diagnosis is suspected through medical history of abnormal fatigability and corroborated by physical examination, repetitive nerve stimulation of an affected but not complete paralyzed muscle, correction of fatigability by the intravenous administration of acetylcholine esterase inhibitors, and by the presence of serum acetylcholine receptors antibodies (ACRA). The long term treatment is symptomatic (acetylcholine inhibitors) and etiopathogenic (immunosupresor drugs, plasmapheresis, intravenous gamma globulin and thymectomy. Spontaneous or post symptomatic and etiopathogenic treatment remissions occur from 1 to 10 years. Fatality is rare but children are at high risk during myasthenia crisis.
Key words: Myasthenia gravis; Diagnosis; Treatment; ACRA
La Miastenia Grave Juvenil (MGJ) es un trastorno crónico auto inmune de la transmisión neuromus-cular que se manifiesta por fatiga muscular precoz con progresión a la parálisis durante estados de contracción muscular iterativos (movimientos) o sostenidos (posturas)1-3. La recuperación de la fuerza muscular ocurre de segundos a minutos después que se interrumpe la contracción muscular. Por lo tanto, los síntomas de fatiga o debilidad muscular son más obvios hacia el final del día. Los músculos extra oculares y elevadores de los párpados tienen más tendencia a la parálisis sin recuperación durante el reposo que los músculos inervados por otros pares craneales y de las extremidades.
La incidencia de MG en los primeros 18 años de vida es de 4 por cada 100,000 habitantes4, 5. La forma adquirida auto inmunológica juvenil es la más frecuente (18%) seguida por la forma adquirida inmunológica transitoria neonatal (1.5%) y congénita (0.5%)5.
Las formas de presentación de la MGJ son generalizadas (47%), ocular (43%) y crisis miasténicas (10%)6. Al cabo de 1-3 años la forma generalizada aumenta al 80%, las formas oculares disminuyen al 20% y las crisis miasténicas, que ocurren solamente en las formas generalizadas, disminuyen al 5%7, 12·.
El diagnóstico clínico se basa en demostrar fatiga precoz progresando a parálisis de los músculos afectados mediante contracción sostenida o repetitiva de los músculos afectados pero no paralizados, durante el reposo, y la recuperación de la contracción muscular al cesar la contracción de estos músculos. El diagnóstico se corrobora si al administrar un inhibidor de la acetilcolinesterasa (IACE) se restituye la función de los músculos afectados al aumentar la concentración de acetilcolinesterasa AC en la hendidura sináptica con activación más sostenida de los receptores de acetilcolin (RAC) disponibles.
El diagnóstico de certeza se realiza mediante la presencia de anticuerpos contra receptores de acetilcolin (ACRA) en el 80% y 50% de las formas generalizada y ocular (MGJ seropositiva)2, 13. El resto no presentan ACRA (MGJ seronegativa). Entre el 31% y 41% de estos pacientes seronegativos tienen anticuerpos contra la proteína kinasa específica muscular (APKEM)14.
La demostración de una disminución de más del 10% de la amplitud del cuarto y quinto potencial de acción muscular compuesto (PAMC), comparado con el primer PAMC evocado mediante la estimulación eléctrica supramáxima repetitiva a 2 Hz por 20 segundos de por lo menos un nervio distal y uno proximal, inervando los músculos afectados pero no paralizados corroboran el diagnóstico sobre todo en las formas generalizadas. La electromiografía de una sola fibra muscular también se emplea en casos dudosos.
El tratamiento es sintomático y a largo plazo con IACE. En las formas oculares puras no ofrecemos tratamiento alguno si los IACE son inefectivos9. En las formas generalizadas auto inmunológicas que no responden completamente a los IACE se añaden numerosas medidas para frenar la respuesta auto inmunológica. Entre éstas se encuentran la timectomía, que sigue siendo la más empleada en pacientes post puberales seropositivos a pesar de que su eficacia no ha sido demostrada15, los agentes inmusupresores como esteroides, azatioprina, ciclofosfamida, ciclosporina, tacrolimo y mofetil micofenolato y la gammaglobulina endovenosa16, 17.
En los pacientes con crisis miasténicas con o sin timectomía previa, la plasmaféresis seguida por gamma globulina intravenosa siguen siendo la mejor elección a pesar de que su eficacia no ha sido documentada en estudios controlados a doble ciego16, 17.
El objetivo de esta revisión es actualizar el conocimiento de la miastenia grave juvenil auto inmunológica principalmente en las áreas de patogénesis, diagnóstico y tratamiento.
Epidemiología
La incidencia de MG es de 2.5-20/100 000 personas, correspondiendo un 20% a los pacientes en las primeras dos décadas de la vida4, 5. La incidencia de las forma juvenil auto inmunológica, neonatal inmunológica y congénita son 18%, 1.5% y 0.5% respectivamente. La MGJ auto inmunológica es más frecuente generalizada (80%) que localizada (20%), aunque hasta el 33% de las formas generalizadas comienzan afectando primero a los músculos extraoculares y 10% debutan como crisis miasténicas.
La MGJ ocurre en forma proporcional en ambos sexos en la edad pre puberal, pero predomina en el sexo femenino en la edad post puberal10-14.
Patogénesis
Anatomía normal de la unión neuromuscular
La unión neuromuscular (UNM) es una sinapsis periférica constituida por 1) la región pre sináptica, 2) la región post sináptica y 3) la hendidura sináptica (Fig. 1).

Fig. 1.- Anatomía de la unión neuromuscular. Figura tomada de la referencia 23.
1) La región pre sináptica está constituida por la terminación en forma de un botón de rosa y libre de mielina de los axones mielinizados de las motoneuronas alfa de las astas anteriores de la médula espinal y el tallo cerebral. Los botones sinápticos contienen vesículas con aproximadamente 10.000 moléculas de acetilcolin (AC) densamente agrupadas y situadas frente a la membrana de los terminales pre sinápticos que contienen gran cantidad de canales de calcio dependientes del voltaje y las zonas activas para la exocitosis de AC en la hendidura sináptica. La AC en estas vesículas está rápidamente disponible. Reservas mayores tienen que ser transportadas de áreas más lejanas de la membrana pre sináptica. Esta área del botón sináptico se encuentra de frente al área de alta densidad de receptores de AC de los pliegues de la membrana post sináptica muscular de la UNM.
2) La región post sináptica está constituida por la membrana de la fibra muscular inervada por el botón sináptico que se pliega para aumentar la superficie de contacto sináptico y contiene densamente empaquetados aproximadamente 12,000 moléculas por μm2 de receptores de la acetilcolin (RAC).
Los RAC son glicoproteínas formadas por 5 sub unidades: 2 sub unidades α idénticas que se unen a la AC y 3 sub unidades diferentes pero homólogas β, γ (pre inervación o embrionario) o ε (post inervación o adulta) con un canal iónico central. Algunos músculos en el adulto expresan la forma embrionaria (músculos extraoculares).
En la membrana post sináptica también existen otras proteínas involucradas en el desarrollo de la UNM y agregación de los RAC. Una de estas proteínas es la tiroxina kinasa específica del músculo (TKEM). La misma se expresa en la forma embrionaria y del adulto semejante a los RAC, predominante en la UNM donde es parte del receptor agrin. Agrin es una proteína sintetizada por las neuronas motoras y secretada en el botón sináptico que, al unirse a la TKEM en la membrana muscular post sináptica promueve y mantiene los RAC agregados en presencia de la rapsina. La rapsina es una proteína en la superficie de la membrana post sináptica necesaria para mantener agrupados los RAC. La rapsina y los RAC están presentes en concentraciones equimolares en la UNM y podrían estar físicamente asociados.
La rapsina produce agregación de otras proteínas en la UNM incluyendo la TKEM. Los ratones sin agrin o TKEM no desarrollan UNM y mueren al nacer debido a debilidad muscular marcada y sus RAC se expresan uniformemente en toda la extensión de la membrana de la fibra muscular y no se agrupan en la región de los pliegues.
3) La hendidura sináptica es el espacio (20 nm) entre el botón sináptico y la membrana muscular post sináptica donde se encuentran la acetilcolin esterasa (ACE), que hidroliza a la AC y otras proteínas y glicoproteinas, cuya función es estabilizar la estructura de la UNM18-22.
TABLA 1.- Clasificacion de la miastenia gravis (MG) según la gravedad de los músculos afectados

TABLA 2.- Clasificación de la MG según los músculos afectados y pronóstico

Función normal de la unión neuromuscular
La función de la UNM es transmitir la información entre las motoneuronas alfa de la médula espinal y el tallo cerebral y el músculo para la producción de contracciones musculares sostenidas (posturas) y continuadas (movimientos). El potencial de acción generado en el montículo axónico de las motoneruronas alfa, al llegar al botón sináptico abre los canales de calcio dependientes del voltaje y la entrada intracelular del mismo desencadena la fusión de las membranas de las vesículas y las pre sinápticas del botón terminal, con exocitosis de 60 a 100 vesículas pre sinápticas con liberación de 60 000 a 100.000 moléculas de AC en la hendidura sináptica. La AC es en parte hidrolizada por la ACE pero una cantidad más que suficiente se une a los RAC, abriendo los canales de sodio dependientes de la unión de la AC y los RAC. El sodio penetra rápidamente dentro de la fibra muscular despolarizando la membrana muscular post sináptica. La despolarización de la membrana muscular post sináptica abre los canales de sodio dependientes del voltaje, con aún mayor entrada de sodio y creando un potencial post sináptico (PPS) de 40 mV, o sea por encima del umbral para desencadenar un potencial de acción muscular propagado (25 mV) en la membrana muscular adyacente a la post sináptica que produce los mecanismos involucrados en la contracción y relajación musculares.
El margen de seguridad (MS) de la transmisión neuromus-cular por lo tanto es alto y representa el voltaje en exceso por encima del umbral necesario para evocar un potencial de acción muscular compuesto (PAMC). El margen de seguridad en condiciones normales es de 15 mV estimulando una vez un axón alfa. El MS es de 5 y 10 mV durante la estimulación repetitiva entre 2 y 5 Hz.
Las reservas de AC para respuestas rápidas se agotan con esta frecuencia de estimulación durante las primeras cuatro estimulaciones, con disminución del calcio intracelular pre sináptico. El MS se recupera entre 5 y 10 segundos de mantenerse la estimulación entre 2 y 5 Hz al acumularse más calcio intracelular y movilizar a la AC para respuesta sostenida. La estimulación entre 10-50 Hz o la contracción muscular sostenida voluntaria por 30- 60 segundos produce acumulación mayor de calcio intracelular y liberación aún mayor de AC con la producción de PPS y PAM mayores entre 5 y 60 segundos después de la estimulación tetánica (facilitación post tetánica). Entre los siguientes 1 a 4 minutos después de la estimulación titánica se produce una disminución del MS debido a reducción de los PPS y PAM que puede durar hasta 10 minutos (exhaustación pos tetánica).
Durante el reposo existe exocitosis producida por acumulación intracelular pre sináptica de calcio, al azar y continua de no más de una vesícula sináptica al mismo tiempo, con liberación de un cuanto o 10 000 moléculas de AC que en parte son hidrolizadas por la ACE y en parte se une, a los RAC abriendo los canales de sodio dependientes de la configuración química, y éstos a su vez abren los canales de sodio dependientes del voltaje, con la despolarización de la membrana post sináptica de la UNM y la creación de potenciales post sinápticos aislados no mayores de 1 mV durando entre 5 y 8 milisegundos e incapaces de evocar un potencial de acción muscular propagado todo o nada. Estos PPS se conocen como potenciales en miniatura (PPSM). Los PPSM al despolarizar la membrana post sináptica facilitan la producción de PPS y por lo tanto de PAM propagados23, 24.
Fisiopatología de la unión neuromuscular
En pacientes con MG el MS está disminuido siendo el valor del PPS cada vez menor a medida que se trata de mantener la contracción muscular.
Como hemos señalado anteriormente, el PPS depende de la cantidad de AC liberada por un potencial de acción de la neurona alfa, la densidad de los RAC, la actividad de la ACE en la hendidura sináptica y la presencia de los canales de sodio dependientes del voltaje en la profundidad de los pliegues post sinápticos.
La reducción del número o de la actividad de los RAC en la UNM disminuye el valor de un PPS que podría ser suficiente durante el reposo o la primera contracción, pero no así durante las contracciones musculares sucesivas que llevan a reducción en la liberación rápida de AC y al PPS a valores por debajo del umbral para evocar PAM. Estos eventos son más notables en los músculos extraoculares, que tienen pliegues post sinápticos menos pronunciados con menor cantidad de RAC y canales de sodio sensibles al voltaje.
La naturaleza auto inmunológica de la MGJ se basa en los siguientes datos: 1) 80-90% de los pacientes tienen IgG anticuerpos séricos contra los RAC, 2) la transferencia placentaria de ACRA de madres con MG auto inmunológica produce MG neonatal transitoria, 3) la inoculación de IgG purificada de pacientes con MG en animales produce síntomas miasténicos, 4) la inmunización de animales con RAC purificados de otras especies desencadena no sólo una respuesta inmunológica con la presencia de anticuerpos contra RAC sino MG experimental, 5) la presencia de infiltrados inflamatorios en los músculos de pacientes miasténicos, 6) los cambios tímicos patológicos caracterizados por expansión de los espacios peri vasculares con infiltrados linfoides y folículos de células B en los centros germinales, 7) la presencia de otros desórdenes auto inmune encontrados en pacientes con MG, tales como enfermedad de Graves y Tiroiditis de Hashimoto 8) la asociación con ciertos subgrupos de HLA B8 y A1, DR3/DW3 y ciertos alelos DQ22.
En los pacientes con MGJ los anticuerpos IgG séricos contra los ACRA, al unirse a éstos producen aceleración de la degradación de los RAC por endocitosis y posterior proteolisis y congregación, bloqueo funcional de la región activa y lesión de la membrana post sináptica en colaboración con el complemento. El resultado final es un bloqueo parcial o total de la transmisión neuromuscular a nivel post sináptico, que provoca desde fatigabilidad hasta parálisis transitoria o permanente, (Fig. 2).
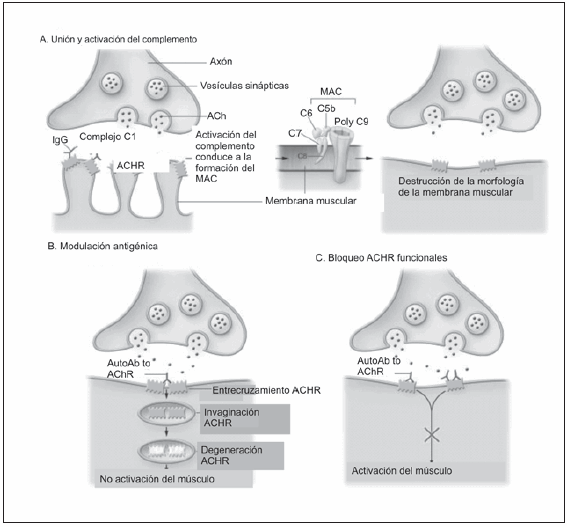
Fig. 2.- Unión y activación del complemento en la unión neuromuscular. Figura tomada de la referencia 23. MAC: Complejos de membrana de ataque. AutoAb to AChr: Autoanticuerpos contra receptores de acetilcolina. ACh: acetilcolina
El mecanismo que inicia y mantiene la respuesta auto inmunológica, produciendo la pérdida de la auto tolerancia, se desconoce. El timo se ha implicado como posible lugar de origen, porque el 75% de los pacientes tienen anormalidades tímicas, un 85% tienen hiperplasia con centros germinales activos y un 15% tienen timomas en mayores de 20 años. La timectomía, por otra parte, mejora a la mayoría de los pacientes.
Los RAC de las células mioides del timo podrían actuar como auto antígenos y poner en marcha la reacción auto inmunológica a nivel del timo21. Los linfocitos del timo (T) estimularían a través de los CD4 a los linfocitos sanguíneos (B), que a su vez iniciarían la producción de anticuerpos contra los RAC. Por lo tanto, el inicio de la producción de anticuerpos contra los RAC se debe a la activación de los linfocitos T en un complejo tri molecular compuesto por las moléculas de histocompatibilidad mayor clase II, los RAC de las células mioides del timo y las células de presentación antigénica.
El rol patogénico de los linfocitos T CD4+ Anti RAC en el desarrollo de los síntomas de MG se basa en la demostración de mejoría tras la timectomía o tras tratamiento con anticuerpos contra CD4, más aún en los pacientes miasténicos con SIDA y disminución de los CD4.
Existe un grupo de pacientes que no presentan en el suero anticuerpos contra los RAC, denominados seronegativos. Recientemente se ha descrito la presencia en el suero de anticuerpos contra la molécula tirosina kinasa muscular específica (TKEM) en un subgrupo de pacientes con MG generalizada seronegativa. Anticuerpos contra la rapsina también han sido identificados en pacientes con MG asi como en aquellos con lupus eritematoso diseminado y miopatía asociada a procainamida. Estos auto anticuerpos contra TKEM se encuentran en el suero de hasta 34.6% de los pacientes con MG seronegativa.
Otros investigadores han reportado cambios tímicos típicos en pacientes con MG seronegativa semejantes a los de MG con ACRA, más frecuentemente hiperplasia tímica, sugiriendo que algunos de estos pacientes pudieran beneficiarse de la timectomía22, 25.
Manifestaciones clínicas
Formas de presentación generalizadas
Las formas generalizadas desde el comienzo ocurren entre el 47-50 % de los niños y adolescentes con MGJ.
Al comienzo la fatiga se ignora ya que nunca llega al extremo de parálisis generalizada, y el diagnóstico es tardío ya que raramente se piensa en esta condición. Los signos de fatiga y debilidad son más evidentes al final del día. Raramente las manifestaciones de fatiga de los músculos inervados por los pares craneales V, VII, IX, X, XI y XII llevan al diagnóstico. El diagnóstico es poe exclusión de causas de decaimiento y cansancio, desde anemia hasta fobia al colegio y actividades físicas.
Ocular
La forma ocular desde el comienzo ocurre entre el 43- 50% de los niños y adolescentes con MGJ, siendo más frecuente en los pre púberes. Los músculos más involucrados son el elevador de los parpados y, de los movimientos de los ojos, sin comprometer al reflejo fotomotor. La fatiga se caracteriza por ptosis y/o diplopía. La ptosis puede ser unilateral o bilateral con distinto grado de afectación y raramente simétrica, y el mismo patrón ocurre con la diplopía, que mayormente es horizontal. Casi siempre el paciente es evaluado por el oftalmólogo cuando la ptosis o la diplopía son persistentes a cualquier hora del día. El oftalmólogo, quien en la ausencia de papiledema y otras manifestaciones de aumento de la presión intracraneal sospecha el diagnóstico, corrobora la fatiga de los músculos extraoculares y elevador del párpado, y hace el diagnóstico en el 85% de los casos con la administración endovenosa de edrofonio en el consultorio en los post púberes y en la sala de urgencias en los pre púberes. Algunos oftalmólogos prefieren no realizar la prueba de edrofonio y determinar la presencia en suero de ACRAC y ATKEM y comenzar con IACE, especialmente piridostigmina oral con fines diagnósticos y terapéuticos. Aproximadamente un tercio de estos pacientes evolucionan hacia la forma generalizada entre 1 y 3 años después del comienzo.
Crisis miasténicas
La forma de comienzo con crisis miasténica es una emergencia médica que ocurre entre el 7 y 10% de los pacientes con MGJ, con más frecuencia en los post púberes.
La fatiga se manifiesta por disnea durante la actividad que desaparece con el reposo hasta que se hace persistente en el curso de 12 a 24 horas acompañada de disfagia, disfonía, sudoración, taquicardia y parálisis de las extremidades con incapacidad para caminar e insuficiencia respiratoria. El paciente es atendido en la escena con ventilación asistida y oxígeno y transportado a la sala de urgencias donde es intubado y admitido a la unidad de cuidados intensivos. La exclusión de otras causas de insuficiencia respiratoria y parálisis generalizada llevan unas horas o días más tarde al diagnóstico de MGJ, que se comprueba mediante la administración endovenosa de edrofonio o neostigmina. Todos estos pacientes, en mi experiencia tienen ACRA elevados y responden favorablemente a los IACE endovenosos y la plasmaféresis seguida de gamma globulina endovenosa por 5 días cada tratamiento.
Se les da de alta con IACE y un inmunosupresor, generalmente prednisona. La recurrencia de crisis miasténicas en estos pacientes es alta y siempre se debe administrar edrofonio antes de comenzar la plasmaféresis para asegurarnos que la crisis no se debe a exceso de IACE.
Curso
La severidad de los síntomas progresa de semana a meses, desde las formas más ligeras a las más severas la debilidad muscular tiende a extenderse desde los músculos oculares a los faciales, bulbares y luego tronco y extremidades. Las enfermedades intercurrentes como las virales o bacterianas respiratorias se identifican como la causa más frecuente que desencadena descompensación y agravamiento de los síntomas, con crisis miasténicas con compromiso respiratorio rápido y severo. Otras condiciones intercurrentes que pueden desencadenar crisis miasténicas son: post cirugía, embarazo, menstruaciones, post parto, medicamentos tales como antibiótico, (aminoglucósidos, tetraciclinas y eritromicinas), antiarrítmicos (propanolol, quinina, lidocaína), antipsicóticos (litio, clorpromazina) antiepilépticos (fenitoina y carbamazepina)5, 13, 15.
En los pacientes miasténicos es usual encontrar otras enfermedades auto inmunológicas tales como hipertiroidismo, artritis reumatoide y lupus eritematoso26, 27.
Diagnóstico
El diagnóstico de MGJ en la mayoría de los pacientes demora entre 3 y 9 meses después del comienzo de los síntomas. Esto se debe a que no se piensa en la condición y a veces los síntomas son tomados muy a la ligera.
El diagnóstico clínico se basa en demostrar fatiga precoz progresando a parálisis de de los músculos mediante contracción sostenida o repetitiva éstos afectados pero no paralizados en forma permanente, y la recuperación de la fuerza muscular al cesar la contracción de estos músculos.
El diagnóstico de certeza se realiza mediante la demostración de ACRA séricos en el 80% y 50% de las formas generalizada y ocular (MGJ seropositiva)14. El resto no presentan ACRA (MGJ seronegativa). Entre el 31% y 41% de estos pacientes seronegativos tienen anticuerpos contra la proteína kinasa específica muscular (ATKEM)15. En el grupo seropositivo ordenamos anticuerpos contra titin y anti estriaciones musculares y tomografía axial computarizada del timo para descartar timoma, pero no realizamos la prueba del cloruro de edrofonio endovenosa ni realizamos las pruebas neurofisiológicas.
En los pacientes seronegativos realizamos la prueba endovenosa de cloruro de edrofonio y solamente si es negativa entonces las pruebas neurofisiológicas y tomografía axial computarizada del timo para descartar timoma.
En los pacientes con MGJ que debutan con crisis, el diagnóstico se hace mediante la administración endovenosa de edrofonio o neostigmina para, si positiva, continuar el tratamiento con IACE y tomamos muestras de suero para ACRA y TKEM antes de realizar la plasmaféresis.
Anticuerpos contra los receptores de acetilcolina (ACRA)
La presencia sérica de ACRA es diagnóstica de MGJ. Mediante la prueba de radioinmunoanálisis se detectan entre un 85% y un 90% de los pacientes con miastenia generalizada y un 50% de los pacientes con formas oculares (seropositivas). Sólo en casos excepcionales de pacientes con lupus eritematoso sistémico se han dado resultados falsos positivos4, 5. Aproximadamente en el 15% de los pacientes con MGJ generalizada no se puede demostrar la existencia de anticuerpos, son los llamados miasténicos seronegativos, los cuales no se diferencian clínicamente ni en la respuesta a los inmunosupresores de los que tienen títulos elevados. Entre el 31% y 41% de estos pacientes seronegativos tienen anticuerpos contra la proteína kinasa específica muscular (ATKEM)14. Algunos pacientes seronegativos no tienen ACRA o ATKEM sino un factor plasmático que activa un mensajero secundario en el músculo que induce la fosforilación e inactivación de los RAC28. Otros pacientes pueden también tener anticuerpos contra proteínas miofibrilares29 tal como aquellos contra la miosina y la treponina de acción rápida que interactúan contra los RAC30. Por último los pacientes con MGJ y timomas, que raramente ocurre por debajo de los 20 años de edad, pueden presentar anticuerpos contra el titin31 y el receptor rianodina32.
Prueba del cloruro de edrofonio endovenoso
El cloruro de edrofonio (CE) intravenoso es preferido a los otros IACE por su rápida acción (1-3 minutos) aunque tiene el inconveniente de una vida corta (5-15 minutos). El objetivo de la prueba es corregir la debilidad muscular al aumentar la concentración de AC en la hendidura sináptica que al unirse a los RAC produce un PPS por encima del umbral para evocar PAM y desencadenar los mecanismos de la contracción muscular en pacientes con MGJ. El procedimiento debe hacerse bajo supervisión médica y control continuado de la presión, pulso y saturación de oxígeno en un sitio con facilidades para atender niños con paro respiratorio o cardíaco. La vía endovenosa se debe obtener antes de iniciar la prueba y mantener su permeabilidad con heparina. Se deben emplear jeringuillas de 1 centímetro cúbico fraccionadas en décimos (tuberculina) e identificados. Una debe contener solución salina fisiológica estéril, otra sulfato de atropina (0.01 mg/kg/dosis) y la tercera con CE (10 mg/cc). Se recomienda grabar el procedimiento en video con sonido. Se debe de antemano seleccionar los músculos fatigables o paralizados, especialmente los extraoculares o elevadores del parpado. El paciente debe estar acostado en posición supina para evitar hipotensión postural.
Se le debe explicar el procedimiento y los efectos indeseable del medicamento por estimulación excesiva de los RAC muscarínicos (bradicardia, hipotensión, secreciones excesivas de saliva y bronquiales, constricción de los bronquios, arritmias y dolor abdominal). Se administra solución salina estéril para descartar el efecto placebo, sobre todo en los adolescentes. Se esperan 3 minutos y se administra un tercio de la dosis total de CE y se espera por 3 minutos. Si la prueba es positiva mediante la corrección de la ptosis o oftalmoparesis se da por terminada, pero si es negativa se procede a administrar otro tercio de la dosis de CE y se espera por 3 minutos; y si positiva se interrumpe la prueba pero si es negativa se termina infundiendo el último tercio de la dosis total de CE y se esperan 3 minutos. Si ocurren síntomas indeseable se administra lentamente atropina. Durante todo el procedimiento se registra continuamente el pulso y la saturación de oxigeno del dedo. La respuesta es positiva cuando se corrigen la ptosis u oftalmoplegia por lo menos por 5 minutos después de administrar el fármaco. La sensibilidad de la prueba es superior al 95%. Se han descrito muy pocos casos de resultado falso positivo al edrofonio en pacientes con enfermedad de neurona motora, síndrome de Guillain-Barré, miastenia congénita, así como falsos negativos en algunas formas miasténicas oculares puras. Pese a que la fiabilidad del diagnóstico no ha sido nunca estudiada, no hay otras enfermedades que presenten una combinación similar de sintomatología clínica, positividad de la prueba de edrofonio y presencia de ACRA.
Estimulación repetitiva de nervio periférico (ERN)
La estimulación eléctrica repetitiva supramáxima a 2 Hz por 5 segundos de al menos dos nervios motores inervando músculos débiles, uno distal y otro proximal, debe producir una disminución de la amplitud del PAM 4 y 5 de al menos el 10%, cuando se compara con la amplitud del primer PAM a fin de considerar la prueba positiva. La disminución de la amplitud del PAM es evidente también entre 2 a 10 minutos después de ejercicio sostenido del músculo en prueba por 30 segundos, o la estimulación a la frecuencia de 50 Hz por 1 segundo (agotamiento postetánico).
Durante los primeros 2 minutos después de esta prueba se observa un aumento de la amplitud de los PAM (facilitación post tetánica). A fin de evitar errores se debe mantener la temperatura cutánea por encima de 32 °C y bien inmovilizada la extremidad y los electrodos de registro y estimulación. La prueba se puede realizar en pacientes despiertos o sedados. La sensibilidad de la prueba nunca se ha determinado pero hasta ahora, en mi experiencia, ha sido positiva en todos los pacientes con pruebas de CE positivas y títulos elevados de RAC24.
EMG de fibra muscular aislada
El electromiograma de fibra aislada es la prueba más sensible para detectar trastornos de la transmisión neuromuscular en vivo. Solamente la ordenamos si la prueba de estimulación repetitiva, el EMG convencional, la prueba de CE y los títulos de ACRA son negativos y aún se sospecha el diagnóstico de MGJ. La prueba se realiza con micro electrodos intramusculares que detectan uno y no más de dos potenciales de acción muscular de una o dos fibras musculares de la misma unidad motora. Los músculos más empleados en mayores de 10 años son los músculos faciales y el extensor index propius. Se requiere gran cooperación del paciente, quien voluntariamente tiene que contraer el músculo bajo prueba hasta aislar los potenciales de acción muscular propagados de dos fibras musculares cercanas y medir el intervalo de tiempo entre las dos. En los casos positivos está prolongado hasta desaparecer cuando el bloqueo es completo debido a que el valor del PPS es menor que el umbral para evocar un PAM. En casos que no cooperan se puede realizar bajo sedación y estimulación eléctrica del nervio del músculo bajo prueba. En más de un 95% de los casos el estudio es positivo, incluso en las formas exclusivamente oculares25.
Estudios complementarios
Existen estudios complementarios, entre los que se destacan los exámenes por imágenes, entre ellos la radiografía de tórax, y la TAC y/o RMI de tórax, con el objetivo de descartar el timoma como probable causante de miastenia.
Se recomienda también el estudio de la función pulmonar con espirometría, para evaluar la respuesta a la terapéutica utilizada, aunque no se realiza como norma en todos los pacientes12.
Diagnóstico diferencial
Los síndromes miasténicos congénitos constituyen un conjunto de enfermedades que por distintos mecanismos comprometen la transmisión neuromuscular, lo cual se expresa clínicamente con debilidad y fatiga. No están originados por un proceso auto inmune, faltando por lo tanto los anticuerpos específicos anti receptor de acetilcolin y no responden al tratamiento inmunosupresor; la mayoría son hijos de madres no miasténicas, siguen un patrón de herencia autosómico recesivo con antecedentes de consanguinidad frecuentes. La mayoría presentan síntomas al nacimiento o los desarrollan durante los dos primeros años de vida. Recientemente se han identificado varios síndromes que se definen por criterios clínicos, electrofisiológicos y morfológicos específicos. Se derivan de alteraciones pre sinápticas, sinápticas o post sinápticas32.
Tratamiento
El tratamiento de elección de la MGJ es etiopatogénico pero desafortunadamente aún no se ha logrado. De manera que no existe cura para la MGJ. Afortunadamente la tasa de remisión espontánea (26%) o con la terapia empleada en estos momentos es alta (76%)16, 26. Mientras tanto el tratamiento es sintomático y dirigido a frenar la destrucción auto inmunológica continua de los RAC.
Inhibidores de la acetilcolin esterasa (IACE)
Los IACE compiten con la AC para unirse a la ACE. De manera que existe un exceso de AC en la hendidura sináptica que al unirse a la AC liberada al azar espontáneamente y refleja, y voluntariamente para mantener posturas y movimientos, genera PPSM y PPS de un voltaje suficiente como para producir PAM propagados.
La dosis tiene que ser ajustada ya que si es excesiva podría agravar los síntomas por bloqueo debido a despolarización excesiva y continua de la membrana post sináptica a nivel de la UNM y la producción de síntomas parasimpaticomiméticos sistémicos como bradicardia, dolor abdominal, diarreas, aumento en lacrimación y secreciones orofaríngeas, comprometiendo aún más la respiración y deglución, conduciendo a una crisis miasténicas por exceso más bien que por déficit de AC. En estos casos la administración endovenosa de otro AC, cloridato de edrofonio, de corta vida y rápida acción, empeoraría transitoriamente los síntomas.
El IACE más empleado en forma oral es el bromuro de piridostigmina, y en forma endovenosa el sulfato de prostigmina.
El bromuro de piridostigmina está disponible en forma de tabletas de acción regular (60 mg) y prolongada (180 mg), y líquido (60 mg/5ml). La dosis de comienzo es de 6.3 mg/kg/24 h dividida cada 3-4 horas mientras el paciente está despierto y posteriormente se ajusta hasta conseguir los objetivos o la aparición de efectos indeseables intolerables. La dosis máxima es 7.7 mg/kg/24 h dividida cada 3-4 horas mientras esté despierto. Se absorbe mal por el tubo digestivo, no atraviesa la barrera hematoencefálica y se elimina inalterada por el riñón. El comienzo de la acción oscila entre 30 y 60 minutos y los efectos duran entre 3 y 6 horas. Preferimos la forma líquida con sabor a ciruela, ya que la podemos fraccionar más fácilmente que la tableta en los pre púberes. Raramente empleamos la forma de acción prolongada. La forma prolongada se administra cada 6-8 horas y equivale a recibir 60 mg cada 3 horas de las tabletas regulares. No se puede masticar o quebrar por el riesgo de una sobredosis.
Los IACE no se deben usar si el paciente sufre obstrucción intestinal o urinaria o peritonitis y con mucha precaución en casos con asma. Tampoco el concomitante de boqueadores musculares despolarizantes para evitar una crisis colinérgica. Los efectos indeseables se deben a la excitación de receptores muscarínicos de AC sistémicos e incluyen sialorrea, cólicos abdominales, visión borrosa e hiperemia cutánea. El antídoto por excelencia en el sulfato de atropina intravenoso (0.01 mg/kg/dosis). La eficacia es completa en el 60% de los pacientes.
El sulfato de prostigmina está disponible en forma líquida estéril para uso endovenoso, intramuscular y subcutáneo (0.5 mg/1 ml). La forma endovenosa se usa en pacientes que no pueden tragar o están bajo ventilación asistida en la unidad de cuidados intensivos. La dosis endovenosa de comienzo es de 0.01 mg/kg/dosis cada 1-2 horas. La dosis máxima es 0.04 mg/kg/dosis cada 1- 2 horas. No traviesa la barrera hematoencefálica. El comienzo de la acción oscila entre 5 y 15 minutes y los efectos duran entre 45 y 60 minutos. No se debe usar si el paciente sufre obstrucción intestinal o urinaria o peritonitis, y hacerlo con mucha precaución en casos con asma. Evitar empleo concomitante de boqueantes musculares despolarizantes para evitar una crisis colinérgica. El antídoto por excelencia en el sulfato de atropina intravenoso (0.01 mg/kg/dosis). Se puede usar simultáneamente en forma profiláctica con cada dosis de prostigmina o entre dosis. La eficacia de este tratamiento es total.
Inhibidores de la expresión de la ACE
La ACE-R es una variante rara de ACE que se observa en pacientes con MGJ y animales con MGAE resistente a los efectos de los inhibidores de la ACE que hidroliza la AC contribuyendo aun más a empeorar la transmisión neuromuscular.
Se ha logrado un oligonucleótido sin sentido, EN101, que suprime la expresión de ACE-R, normalizando la transmisión neuromuscular en la MGAE al modular la síntesis de las variantes de ACE y por consiguiente la velocidad de hidrólisis y la eficacia de la activación de los RAC. La eficacia del EN101 en humanos está en investigación33.
Inhibidores de la destrucción de receptores de acetilcolina
Inmunosupresores
Entre los inmusupresores, los más empleado son los esteroides y dentro de ellos la prednisona y la metilprednisolona. Si estos son ineficaces o los efectos indeseables tóxicos, se emplean la azatioprina, ciclofosfamida, ciclosporina, tacrolimo y mofetil micofenolato.
Prednisona/Prednisolona
Las indicaciones son pacientes refractarios a los IACE.
La dosis de comienzo es de 1 mg/kg/24 h dividida en dos dosis y posteriormente se ajusta hasta conseguir los objetivos o la aparición de efectos indeseables intolerables. La dosis máxima es 2 mg/kg/24 h dividida en dos dosis. La mejoría ocurre entre 1-3 meses y una vez logrado el objetivo se mantiene la dosis como mínimo por 6 meses, a no ser que los efectos indeseables dicten reducir la dosis antes o descontinuar el medicamento. Una vez bajo control se puede disminuir gradualmente la dosis a la velocidad de 0.5 mg/kg/semana. Si no se logra la mejoría esperada en 3 meses la MGJ se considera refractaria. Lo mismo sucede en los pacientes que responden favorablemente con dosis que producen toxicidad.
Los esteroides no se deben usar si el paciente sufre infecciones virales, por hongos, o tuberculosis, y con mucha precaución en casos con tendencia a sangrado gastrointestinal. El empleo concomitante de bloqueantes musculares despolarizantes se debe evitar para evitar una crisis colinérgica. Los efectos indeseables incluyen retención de agua y sodio, aumento del apetito y peso, hipertensión arterial, disminución del crecimiento, sangrado gastrointestinal y facie redondeada e hipokalemia. La eficacia es completa en el 60% de los pacientes34, 35.
Azatioprina
La azatioprina está disponible en forma de tabletas de acción regular (50 mg) Las indicaciones son pacientes refractarios a los IACE y esteroides, y en pacientes timectomizados con o sin prednisona o prednisolona previa.
La dosis de comienzo es de 1 mg/kg/24 h dividida en dos dosis y posteriormente se ajusta hasta conseguir los objetivos o la aparición de efectos indeseables intolerables. La dosis máxima es 2 mg/kg/24 h dividida en dos dosis. La mejoría ocurre entre 6-12 meses, y una vez logrado el objetivo se mantiene la dosis como mínimo por 12 meses a no ser que los efectos indeseables dicten reducir la dosis antes o discontinuar el medicamento. Una vez bajo control, se puede disminuir gradualmente la dosis a la velocidad de 0.5 mg/kg/semana. Si no se logra la mejoría esperada en 12 meses la MGJ se considera refractaria. Lo mismo sucede en los pacientes que responden favorablemente con dosis que producen toxicidad.
La azatioprina no se debe usar si el paciente desarrolla síntomas de gripe. Los efectos indeseables incluyen anorexia, náusea, vómitos, infecciones oportunistas, trombocitopenia y leucopenia. La eficacia es completa en una minoría de pacientes (20%)36.
Ciclosporina
Las indicaciones son pacientes refractarios a los IACE y esteroides, y en pacientes timectomizados con o sin prednisona o prednisolona previa.
La dosis de comienzo es de 4 mg/kg/24 h dividida en dos dosis y posteriormente se ajusta hasta conseguir los objetivos o la aparición de efectos indeseables intolerables. La dosis máxima es 8 mg/kg/24 h dividida en dos dosis. La mejoría ocurre entre 1-3 meses y una vez logrado el objetivo se mantiene la dosis como mínimo por 12 meses a no ser que los efectos indeseables dicten reducir la dosis antes o descontinuar el medicamento. Una vez bajo control por 6 meses, se puede disminuir gradualmente la dosis a la velocidad de 0.5 mg/kg/semana. La dosis de mantenimiento es la menor con la cual podamos mantener el paciente libre de síntomas. La duración es por lo menos de 1 año. Si no se logra la mejoría esperada en 12 meses la MGJ se considera refractaria. Lo mismo sucede en los pacientes que responden favorablemente con dosis que producen toxicidad.
La ciclosporina no se debe usar si el paciente desarrolla síntomas de toxicidad renal. Los efectos indeseables incluyen hipertensión arterial, anorexia, nausea, vómitos, infecciones oportunistas, trombocitopenia y leucopenia. La tasa de remisión con este medicamento se desconoce37.
Tacrolimo
El Tracolimo o FK 506 suprime la producción de interleukina- 2 relacionada con la activación de los linfocitos T, inhibiendo la diferenciación y proliferación de los linfocitos T citotóxicos y reduce los ACRA séricos. Las indicaciones son pacientes refractarios a los IACE y esteroides, y en pacientes timectomizados con o sin prednisona o prednisolona previa.
La dosis de comienzo es de 1 mg/kg/24 h dividida en dos dosis y posteriormente se ajusta hasta conseguir el objetivo basado en la determinación de valores séricos pre dosis entre 7 y 8 ng/ml o la aparición de efectos indeseables intolerables. La dosis máxima es 5 mg/kg/24 h dividida en dos dosis. La mejoría ocurre entre 1-3 meses y una vez logrado el objetivo se mantiene la dosis como mínimo por 12 meses, a no ser que los efectos indeseables dicten reducir la dosis antes o discontinuar el medicamento. Una vez bajo control por 6 meses, se puede disminuir gradualmente la dosis a la velocidad de 0.5 mg/kg/semana. La dosis de mantenimiento es la menor con la cual podamos mantener el paciente libre de síntomas. La duración es por lo menos 1 año. Si no se logra la mejoría esperada en 12 meses la MGJ se considera refractaria. Lo mismo sucede en los pacientes que responden favorablemente con dosis que producen toxicidad.
El tacrolimo no se debe usar si el paciente desarrolla síntomas de toxicidad renal. Los efectos indeseables incluyen hipertensión arterial, anorexia, nausea, vómitos, infecciones oportunistas, trombocitopenia, y leucopenia, temblores, cefaleas, diarrea, insomnio, o pinchazos en las manos y los pies. La tasa de remisión con este medicamento se desconoce4, 39.
Mofetil micofenolato
El mofetil micofenolato está disponible en forma de tabletas y líquido. Su eficacia no ha sido demostrada en estudios controlados a doble ciego40. Su toxicidad es menor que los otros inmunosupresores. Actúa inhibiendo selectivamente la actividad de la síntesis de la guanosina en las células T y B activadas41. Los estudios a doble ciego y controlados aun no han concluido su eficacia.
Otros agentes menos empleados en MGJ
Se emplean solamente en casos refractarios a los esteroides o los inmunosupresores anteriormente señalados. Entre ellos se encuentran la ciclofosfamida en combinación con el transplante de medula ósea42 o con rituximab (10 mg/ml), un anticuerpo monoclonal contra el marcador CD20 en la superficie de las células B43.
El Etanercept, un recombinante soluble de los receptores TNF que competitivamente bloquea la acción del TNF-α, es efectivo en pequeñas series44.
Plasmaféresis
La plasmaféresis consiste en reemplazar de 1-1.5 veces el volumen de plasma con solución salina, albúmina, o fracción de la proteína plasmática a fin de reducir los títulos de ACRA séricos45. En los pacientes con crisis miasténicas con o sin timectomía previa, la plasmaféresis seguida por gamma globulina intravenosa siguen siendo la mejor elección, a pesar de que su eficacia no ha sido documentada en estudios controlados a doble ciego.
Se ha desarrollado una técnica más sofisticada para eliminar los ACRA empleando métodos de inmunoabsorción46.
Terapia intravenosa con gamma globulina
La terapia con gamma globulina endovenosa consiste en administrar por 5 días a la dosis de 0.4 g/kg/día. El mecanismo de acción es complejo e incluye inhibición de las citokinas, competencia con auto anticuerpos, inhibición de la deposición del complemento, interferencia con la unión de receptores Fc en los macrófagos y receptores Ig en las células B, e interferencia con el reconocimiento de antígenos por las células T sensibilizadas47.
Innovaciones
La vacuna contra la MGJ48 y los anticuerpos monoclonales contra los anticuerpos contra CD449.
Timectomía
La timectomía está indicada en pacientes post puberales después de un año de tratamiento con IACE y esteroides y en pacientes con crisis miasténicas frecuentes con títulos elevados de ACRA. La vías quirúrgicas son trans-cervical, transesternal y la más reciente mediante toracotomía lateral con remoción del timo bajo visión indirecta-directa. Esta última tiene menor morbilidad, el tiempo postoperatorio se reduce a 2 días y la incidencia de crisis miasténicas postoperatorias es nula con la administración pre operatoria de plasmaféresis y los ACRA disminuyen en suero50. La eficacia no ha sido comparada con las otras dos técnicas. Nuestra experiencia es buena. La remisión total ocurre entre el 35 y 65% de los pacientes post púberes al cabo de los 3 años y asciende hasta el 85% al término de los 5 años. El resto de los pacientes mejoran y la mayoría pueden ser manejados con IACE solamente después de los 5 años post timectomía51. Desafortunadamente no existen estudios controlados a doble ciego comparando la eficacia de la timectomía con otras formas terapéuticas52.
Conflicto de interés: Los autores no declaran ningún conflicto de interés.
1. Nastuk WL, Strauss AJ, Osserman KE. Search for a neuromuscular blocking agent in the blood of patients with myasthenia gravis. Am J Med 1959; 26: 394-409. [ Links ]
2. Simpson JA. Myasthenia gravis, a new hypothesis. Scott Med J 1960; 5: 419-36. [ Links ]
3. Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptor. Science 1973; 180: 871-87. [ Links ]
4. Robertson DN. Enumerating neurology. Brain 2000; 123: 663-4. [ Links ]
5. Ponsetia JM, Espina E, Armengola M. Diagnóstico y Tratamiento de la Miastenia grave. Med Clin (Barc) 2000; 115: 264-70. [ Links ]
6. Teng P, Osserman KE. Studies in Myasthenia Gravis. Neonatal and juvenile types. J Mt Sinai Hosp 1956; 23: 711-27. [ Links ]
7. Arroyo H. Myasthenia gravis in childhood and adolescence. Rev Neurol 1996; 24: 1385-9. [ Links ]
8. Haliloglu H. Gender prevalence in childhood multiple sclerosis and myasthenia gravis. J Child Neurol 2002; 17: 390-2. [ Links ]
9. Davitt BV, Fenton GA, Cruz OA. Childhood myasthenia. J Pediatr Ophthalmol Strabismus 2000; 37: 5-14. [ Links ]
10. Morita MP, Gabbai AA, Oliveira AS, Penn AS: Myasthenia gravis in children: analysis of 18 patients. Arq Neuropsiquiatr 2001; 59: 681-5. [ Links ]
11. Garófalo N. Miastenia grave en la infancia. Presentación de 12 casos. Rev Neurol 2002; 34: 908-11. [ Links ]
12. Schmidt NS, Salinas CME, Erazo TR. Miastenia gravis en pediatría. Rev Chil Pediatr 2005; 76; 291-298. [ Links ]
13. Drachman DB. Myasthenia gravis. N Engl J Med 1994; 330: 1797-810. [ Links ]
14. Vincent A, McConville J, Farrugia ME, Newsom-Davis J. Seronegative myasthenia gravis. Semin Neurol 2004; 24: 125-33. [ Links ]
15. Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidencebased review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000; 55: 7-15. [ Links ]
16. Andrews PI: Autoimmune myasthenia gravis in childhood. Semin Neurol 2004; 24: 101-10. [ Links ]
17. Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve 2004; 29: 484-505. [ Links ]
18. Hughes BW, Kusner LL, Kaminski HJ. Molecular architecture of the neuromuscular junction. Muscle Nerve 2006; 33: 445-61. [ Links ]
19. Horton RM, Manfredi AA, Conti-Tronconi B. The 'embryonic' gamma subunit of the nicotinic acetylcholine receptor is expressed in adult extraocular muscle. Neurology 1993; 43: 983-6. [ Links ]
20. Ruegg MA, Bixby JL. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci 1998; 21: 22-7. [ Links ]
21. Glass DJ, DeChiara TM, Stitt TN, DiStefano PS, Valenzuela DM, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation and is a functional receptor for agrin. Cold Spring Harb Symp Quant Biol 1996; 61: 435-44. [ Links ]
22. Marx A, Wilish A, Schultz A, et al: Pathogenesis of myasthenia gravis. Virchows Arch 1997; 430: 355-64. [ Links ]
23. Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia Gravis: Past, present and future. J Clin Invest 2006; 116: 2843-54. [ Links ]
24. Harper, CM. Neuromuscular transmission disorders in childhood. In: Jones, HR, Jr. Bolton, CF, Harper M. Pediatric Clinical Electromyography 1996; 353-85. [ Links ]
25. Romi F, Gilhus NE, Aarli JA. Myasthenia gravis: clinical, immunological, and therapeutic advances. Acta Neurol Scand 2005: 111: 134-41. [ Links ]
26. Rodríguez M, Gómez MR, Howard FM, Taylor WF: Myasthenia gravis in children. Long term follow up. Ann Neurol 1983; 13: 504-10. [ Links ]
27. Plested CP, Tang T, Spreadbury I, et al. AChR phosphorylation and indirect inhibition of AChR function in seronegative MG. Neurology 2002; 59: 1672-3. [ Links ]
28. Romi F, Skeie GO, Gihus NE, Aarli JA. Striational antibodies in myasthenia gravis: reactivity and possible clinical significance. Arch Neurol 2005; 62: 442-6. [ Links ]
29. Mohan S, Barohn RJ, Jackson CE, Krolick KA. Evaluation of myosin-reactive antibodies from a panel of myasthenia gravis patients. Clin Immunol Immunopathol 1994; 70: 266-273. [ Links ]
30. Aarli J.A. Titin, thymoma, and myasthenia gravis. Arch Neurol 2001; 58: 869-70. [ Links ]
31. Baggi F, Andreetta F, Antozzi C et al. Anti-titin and antiryanodine receptor antibodies in myasthenia gravis patients with thymoma. Ann N Y Acad Sci 1998; 841: 538-41. [ Links ]
32. Bromberg, M.B. Myasthenia gravis and myasthenic syndromes. In: Motor disorders. D.S. Younger, editor. Philadelphia. Lippincott Williams & Wilkins 2005; p 231-46. [ Links ]
33. Brenner T, Hamra-Amitay Y, Evron T, Boneva N, Seidman S, Soreq H. The role of readthrough acetylcholinesterase in the pathophysiology of myasthenia gravis. FASEB J 2003; 17: 214-22. [ Links ]
34. Bedlack RS, Sanders DB. Steroid treatment for myasthenia gravis: steroids have an important role. Muscle Nerve 2002; 25: 117-21. [ Links ]
35. Lindner A, Schalke B, Toyka KV. Outcome in juvenile-onset myasthenia gravis: a retrospective study with long-term follow-up of 79 patients. J Neurol 1997; 244: 515-20. [ Links ]
36. Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group Neurology 1998; 50: 1778-83. [ Links ]
37. Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med 1987; 316: 719-24. [ Links ]
38. Tindall RS, Phillips JT, Rollins JA, Wells L, Hall K. A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci 1993; 681: 539-51. [ Links ]
39. Ponseti JM, Azem J, Fort JM, Codina A, Montoro JB, Armengol M. Benefits of FK506 (tacrolimus) for residual, cyclosporin- and prednisone-resistant myasthenia gravis: one-year follow-up of an open-label study. Clin Neurol Neurosurg 2005;107: 187-90. [ Links ]
40. Ciafaloni E. Mycophenolate mofetil and myasthenia gravis. Lupus 2005; 14 (Suppl. 1): s46-s49. [ Links ]
41. Chaudhry V, Cornblath DR, Griffin JW, O'Brien R, Drachman DB. Mycophenolate mofetil: a safe and promising immunosuppressant in neuromuscular diseases. Neurology 2001; 56: 94-6. [ Links ]
42. Drachman DB, Jones RJ, Brodsky RA. Treatment of refractory myasthenia: "rebooting" with high-dose cyclo-phosphamide. Ann Neurol 2003; 53: 29-34. [ Links ]
43. Pescovitz M.D. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am J Transplant 2006; 6: 859-866. [ Links ]
44. Tuzun E, Meriggioli MN, Rowin J, Yang H, Christadoss P. Myasthenia gravis patients with low plasma IL-6 and IFN-gamma benefit from etanercept treatment. J Autoimmun 2005; 24: 261-8. [ Links ]
45. Newsom-Davis J. Therapy in myasthenia gravis and Lambert-Eaton myasthenic syndrome. Semin Neurol 2003; 23: 191-8. [ Links ]
46. Psaridi-Linardaki L, Trakas N, Mamalaki A, Tzartos SJ. Specific immunoadsorption of the autoantibodies from myasthenic patients using the extracellular domain of the human muscle acetylcholine receptor alpha-subunit. Development of an antigen-specific therapeutic strategy. J Neuroimmunol 2005; 159: 183-191. [ Links ]
47. Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science 2001; 291: 484-6. [ Links ]
48. Cohen-Kaminsky S, Jambou F. Prospects for a T-cell receptor vaccination against myasthenia gravis. Expert Rev Vaccines 2005; 4: 473-92. [ Links ]
49. Ahlberg R, et al. Treatment of myasthenia gravis with anti-CD4 antibody: improvement correlates to decreased T-cell autoreactivity. Neurology 1994; 44: 1732-7. [ Links ]
50. Kumar A. Thoracoscopic thymectomy for juvenile myasthenia gravis. Indian Pediatr 2002; 39: 1131-7. [ Links ]
51. Popescu I. Thymectomy by thoracoscopic approach in myasthenia gravis. Surg Endosc 2002; 16: 679-84. [ Links ]
52. Jaretzki A., Steinglass K.M., Sonett J.R. Thymectomy in the management of myasthenia gravis. Semin Neurol 2004; 24: 49-62. [ Links ]

