










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMedicina (Buenos Aires)
versão On-line ISSN 1669-9106
Medicina (B. Aires) vol.70 no.1 Ciudad Autónoma de Buenos Aires jan./fev. 2010
ARTÍCULO ESPECIAL
Rol de las células T regulatorias en esclerosis múltiple
Juan Ignacio Rojas1, Sergio J. González2, Liliana Patrucco1, Edgardo Cristiano1
1Sección de Neuroinmunología y Enfermedades Desmielinizantes, Servicio de Neurología,
2Unidad de Medicina Molecular y Genómica, Instituto de Ciencias Básicas y Medicina Experimental, Hospital Italiano de Buenos Aires
Dirección Postal: Dr. Juan Ignacio Rojas, Servicio de Neurología, Hospital Italiano de Buenos Aires, Gascón 450,1181 Buenos Aires, Argentina Fax: (54-11) 4959-0322 e-mail: juan.rojas@hospitalitaliano.org.ar
Resumen
La esclerosis múltiple (EM) es una enfermedad inflamatoria autoinmune desmielinizante del sistema nervioso central (SNC). La mayoría de las enfermedades autoinmunes se originan por la activación anormal de la respuesta inflamatoria contra auto-antígenos (la mayoría de ellos desconocidos a la fecha) como consecuencia de la pérdida de la tolerancia periférica. Las células T-regulatorias constituyen un grupo esencial de linfocitos T encargados del mantenimiento de la tolerancia periférica, la prevención de enfermedades autoinmunes y la limitación de enfermedades inflamatorias crónicas. Teniendo en cuenta la importancia de la tolerancia periférica, las células T-regulatorias serían componentes cruciales en el escenario fisiopatológico de los procesos autoinmunes, incluyendo la EM. El presente trabajo recopila los conocimientos actuales sobre la función de las células T-regulatorias en la EM, la enfermedad autoinmune desmielinizante del SNC más prevalente en los seres humanos.
Palabras clave: Células T regulatorias; Enfermedades desmielinizantes; Esclerosis múltiple; Encefalomielitis autoinmune experimental
Abstract
Role of T-regulatory cells in multiple sclerosis. Multiple sclerosis (MS) is an autoimmune inflammatory demyelinating disease of the central nervous system (CNS). Most of autoimmune diseases arise by an abnormal activation of the inflammatory response against self-antigens (most of them unknown up to date) as a consequence of dysfunction in peripheral tolerance. Regulatory T-cells are essential for maintaining peripheral tolerance, preventing autoimmune diseases and limiting chronic inflammatory conditions. Based on that knowledge, T-regulatory cells have emerged as a key component of the physiopathology of autoimmune diseases including MS. This review compiles the current knowledge on the role and function of T-regulatory cells in MS, the most prevalent CNS autoimmune disease in humans.
Key words: T-regulatory cells; Demyelinating disease; Multiple sclerosis; Experimental autoimmune encephalomyelitis
La falta de respuesta del sistema inmune (SI) a la estimulación antigénica se conoce como tolerancia inmunológica. La capacidad del SI de mantener la tolerancia frente a antígenos propios se conoce como autotolerancia.
La pérdida de la auto-tolerancia da lugar a reacciones inmunitarias contra antígenos propios, originándose de esta forma las enfermedades autoinmunes1.
El proceso de tolerancia puede ser inducido en varios estadios del desarrollo y de la activación linfocitaria, pudiendo así identificarse una tolerancia central y una tolerancia periférica. La tolerancia central es inducida por los órganos linfoides generadores (timo y médula ósea). En estos órganos, los antígenos presentes son en su mayoría propios. Los clones de linfocitos cuyos receptores son específicos y que se unen con gran afinidad a estos antígenos propios son eliminados. Esto asegura que los tejidos periféricos se encuentren libres de linfocitos autorreactivos al ser eliminados en los órganos linfoides generadores1. Este proceso se conoce como selección negativa.
La tolerancia periférica consiste en la inducción a una falta de respuesta inmunológica fuera de los órganos linfoides generadores, frente al encuentro de los linfocitos maduros autorreactivos con antígenos propios1. Los principales mecanismos a través de los cuales se induce la tolerancia periférica son: a) eliminación o deleción del clon de linfocitos T (LT) autorreactivos mediante un proceso de muerte celular inducida por la activación; b) anergia clonal o inactivación funcional de los LT autorreactivos sin muerte celular, por reconocimiento de antígenos propios sin coestimulación; c) supresión de la activación de los LT autorreactivos mediada por la actividad de un grupo de linfocitos T conocidos como células T-regulatorias (T-regs). Estas células constituyen una herramienta central del SI en el mecanismo de tolerancia periférica, al bloquear la activación de los LT autorreactivos que reaccionan contra antígenos propios.
El fracaso o la ruptura de los mecanismos previamente expresados de tolerancia, tanto central como periférica, dan origen a los procesos y enfermedades autoinmunes.
Las enfermedades autoinmunes del sistema nervioso central (SNC) se manifiestan fundamentalmente como procesos desmielinizantes. La enfermedad desmielinizante autoinmune más prevalente en seres humanos es la esclerosis múltiple (EM)2, siendo ésta el prototipo de enfermedad inflamatoria autoinmune del SNC3, 4 . La EM afecta a individuos entre los 18 y los 35 años, caracterizándose histopatológicamente por la presencia de placas inflamatorias en la sustancia blanca cerebral y daño axonal5. La desmielinización en la enfermedad está mediada por una respuesta inflamatoria que involucra LT CD4+, CD8+ y macrófagos, siendo el daño causado por un mecanismo de hipersensibilidad tipo IV1. Su etiopatogenia estaría relacionada a una falla en la tolerancia central y periférica. Recientes avances en la investigación han dirigido su atención al papel de las T-regs en la falla de la tolerancia periférica y consecuente génesis de la EM, priorizando esta disfunción sobre la falla de los mecanismos reguladores de la tolerancia central5-7.
Estos hallazgos abren un nuevo campo de investigación en las enfermedades autoinmunes, no sólo de su etiofisiopatología sino también en el aspecto terapéutico.
El objetivo del presente trabajo fue revisar el rol de las T-regs en el desarrollo de la EM.
Subtipos de T-regs
Existen diversos informes que demuestran que la población de T-regs no sería homogénea, habiéndose identificado al menos dos poblaciones de T-regs que diferirían desde el desarrollo en sí, por su fenotipo y por su actividad8.
Estas dos poblaciones de T-regs son sub-clasificadas en T-regs naturales y T-regs inducidas o adaptativas. La mayoría de las T-regs naturales expresan constitutivamente el receptor de IL-2 o la cadena CD25. Estas células son producidas por el timo como una población madura, siendo su desarrollo y función dependientes de la expresión del factor de trascripción Foxp3 (forkhead box p3)9.
La producción de células T-regs adaptativas es inducida a partir de LT vírgenes mediante la estimulación antigénica específica10 durante un proceso inflamatorio en los tejidos periféricos. Es decir que, al producirse un proceso inflamatorio, las citoquinas circundantes y el medio inflamatorio inducen a un grupo de LT a adoptar las características de células T-regs. Es posible inducir in vitro el desarrollo de células T-regs adaptativas provenientes de LT vírgenes, tras la estimulación de los mismos con IL-10 y factor transformador de crecimiento beta (TGF-β) en un medio de cultivo adecuado10, 11.
Foxp3 en el desarrollo de las T-regs
Las T-regs naturales expresan el factor de transcripción Foxp3, un miembro de la familia forkhead/winged9. Foxp3 es un regulador maestro del desarrollo y función de los T-regs. La mutación del gen en humanos es causal de la enfermedad genética IPEX (desregulación inmune, poliendocrinopatía, enteropatía, asociada al cromosoma X). Esta mutación fue observada antes del reconocimiento de la importancia del gen Foxp3 en la regulación de la actividad de los T-regs.
La traducción de Foxp3 induce a los LT vírgenes a aumentar la expresión de CD25 y otras moléculas de superficie asociadas al T-reg tales como CTLA-4 y GITR (glucocorticoid induced TNF receptor family related gene protein), mientras que reduce la producción de IL-2, IFN y e IL-49, 12.
Investigaciones recientes revelan que las células que expresan Foxp3 aparecen poco después del nacimiento, y que el desarrollo de autoinmunidad e inflamación sigue a su depleción13. De esta manera, el factor de transcripción Foxp3 es crítico en los LT TCR + para su diferenciación en T-regs en el timo9.
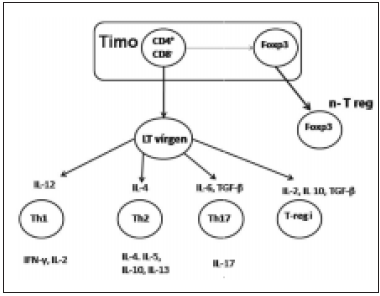
Fig. 1.- Diferenciación de células T vírgenes en células efectoras y origen de las células T-regs.
n-T reg: célula T-reg natural; T-reg i: célula T reg inducida periféricamente; LT: linfocito T.
Cómo ejercen su función supresora las T-regs
Desde la perspectiva funcional, los mecanismos de supresión utilizados por las T-regs pueden ser agrupados en: 1) supresión mediada por citoquinas inhibitorias, 2) supresión mediada por citólisis, 3) supresión mediante disrupción metabólica, 4) supresión mediante la modulación de la maduración y función de las células dendríticas (CPA)14 (Fig. 2).
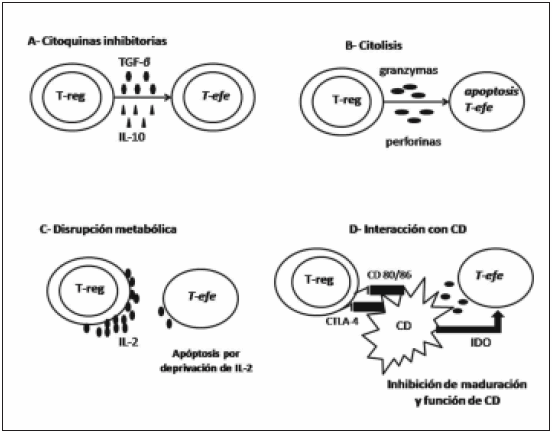
Fig. 2.- Mecanismo de acción de los T-regs. T-efe: linfocito T efector; IDO: indolamina 2-3 dioxigenasa
a) Supresión mediada por citoquinas inhibitorias
Las citoquinas inhibitorias tales como IL-10 y TGF-β han sido el foco de atención como mediadores de la supresión de la respuesta inmune mediada por T-regs14. Varios estudios in vivo soportan esta teoría15. Un ejemplo de ello es el requerimiento de IL-10 para el control de la colitis ulcerosa y el mantenimiento homeostático del número de LT por las T-regs en modelos murinos. En modelos knockout para la producción de IL-10, las células T-regs CD4+ CD25+ fallan en prevenir el desarrollo de colitis ulcerosa, mostrando de esta manera la importancia de estas citoquinas inhibitorias en la supresión mediada por T-regs16.
La expresión de membrana de TGF-β ha sido observada en células T-regs humanas y murinas. Esta expresión de TGF-β en superficie de las T-regs ha sido reproducida tras la estimulación celular con altas dosis de anticuerpos anti CD3. El TGF-β de membrana del T-reg mediaría la supresión de los LT efectores activados a través del contacto célula-célula. Así mismo, el TGF-β de membrana estaría involucrado también en la supresión de las células natural killer (NK)17.
b) Supresión mediada por citólisis
La citólisis por la secreción de granzimas y perforinas ha sido descripta y estudiada ampliamente en células NK y CD8+ citotóxicos. Sin embargo, varios LT CD4+ exhiben una actividad citotóxica14. En relación a esto, se han identificado sub-poblaciones de T-regs que expresan granzimas y estas T-regs han mediado, en modelos murinos y humanos in vitro, la lisis de LT activados mediante el uso de perforinas y granzimas a través de la adhesión vía CD1818,19.
Recientemente se ha demostrado en modelos humanos in vitro que las T-regs suprimen la capacidad de los NK y CD8+ citotóxicos para eliminar células tumorales utilizando el mecanismo de las perforinas y granzimas19.
c) Supresión por disrupción metabólica
Diversos mecanismos de supresión recientemente descriptos han sido propuestos como capaces de producir una disrupción metabólica de los LT efectores14.
Este mecanismo de disrupción estaría dado por el aumento exponencial del marcador CD25 en la superficie de las T-regs. Este aumento de CD25 consumiría la IL-2 circundante necesaria para la proliferación y desarrollo de los LT efectores. Esta depleción de IL-2 conduciría a la apoptosis de los LT efectores20, 21.
Sin embargo, recientes trabajos en T-regs humanos sugieren que la depleción de IL-2 por sí sola no sería un mecanismo por el que las T-regs podrían suprimir al efector22.
d) Supresión mediada por la modulación de la maduración y la función de las células dendríticas
Mas allá del efecto directo de las T-regs en la función de las LT, los mismos podrían modular la maduración y la CPA, necesarias para la activación del LT efector14.
Estudios de microscopía han mostrado una interacción célula-célula entre las T-regs y las CPA. Esta interacción atenuaría la activación del LT efector por parte de la CPA23 en un proceso que involucraría a la molécula co-estimulatoria inhibitoria CTLA-4, expresada constitutivamente en las Tregs24, 25. La expresión de CTLA-4 está aumentada en las T-regs tras la estimulación. Más específicamente, el uso de anticuerpos monoclonales anti CTLA-4 específicos que bloquean la función de esta molécula disminuye la supresión del LT efector mediada por las T-regs12, 26, produciendo enfermedades autoinmunes órgano específicas en ratones, similar a la observada por depleción de T-regs9.
Estos resultados sugieren varias posibles funciones para CTLA-4 en la supresión mediada por las T-regs. Una es que CTLA-4 podría interactuar con CD80 y CD86 en la CPA y traducir una señal co-estimulatoria a las T-regs activándolos y generando la cascada supresora15.
Otro rol posible de CTLA-4 sería generar una supresión directa, ya que CTLA-4 gatillaría la inducción y activación de la enzima indolamina 2-3 dioxigenasa (IDO) en la CPA al interactuar con CD80 y CD8627. IDO catalizaría la conversión de triptofano en leunurenina y otros metabolitos, los cuales tienen un potente efecto inmunosupresor en el ambiente local de las CPA a través de un mecanismo citotóxico, o posiblemente induciendo la generación de T-regs de novo de células T CD25- CD4+27.
T-regs y EM
El SNC ha sido considerado un órgano privilegiado para el desarrollo de la respuesta inmunológica28. Este privilegio hace referencia a que habría cierta protección del tejido cerebral a las respuestas inmunes intraparenquimatosas.
Diversos mecanismos confluyen para otorgar este privilegio inmunológico: 1) la barrera hematoencefálica que ha demostrado prevenir el tráfico de linfocitos al SNC, permitiendo sólo el paso de aquellos linfocitos activados29; 2) se asume que la respuesta inmune podría no desarrollarse en el SNC ya que algunas pocas células residentes expresan moléculas del complejo mayor de histocompatibilidad (CMH) en estado normal30; 3) en el SNC existirían mecanismos locales toleragénicos, específicamente la expresión del ligando de Fas, TGF-β y galectina 9, todos mecanismos asociados con el silenciamiento de los LT ingresados31.
Sin embargo, varias líneas de evidencia indican hoy que esa capacidad del SNC no sería del todo efectiva.
Primero, el acceso de células inmunes al SNC está limitado pero no prohibido. Estudios de migración de LT en ratones sugieren que tanto CD8+ como CD4+ vírgenes son capaces de migrar al SNC31. Segundo, la presentación de antígenos ocurre en el SNC. Luego de la exposición a un ambiente pro-inflamatorio, los oligodendrocitos y las neuronas expresan moléculas de clase I del CMH permitiendo la presentación de antígenos a LT CD8+, mientras que los astrocitos y las células de la microglia expresan moléculas de clase I y II del CMH. Así mismo se ha demostrado también que las CPA residentes serían capaces de activar a los LT CD4+32, 33.
En resumen, aunque en condiciones estables el SNC no favorece la respuesta inmune, nuevos aportes indican que cuando se altera mínimamente ese estado, los LT pueden migrar y desarrollar una respuesta inmune28 .
Control regulatorio de los LT autorreactivos: función del timo en la tolerancia a autoantígenos del SNC
Como mencionamos previamente, la selección negativa de los LT autorreactivos en el timo es un paso clave en la prevención de procesos autoinmunes.
Una variedad de autoantígenos del SNC, relevantes para el desarrollo de enfermedades autoinmunes, son expresados en el timo con el fin de generar la correcta selección negativa de los LT autorreactivos. Estos antígenos son principalmente la proteína básica de mielina, la proteína proteolipídica y la glucoproteína de mielina del oligodendrocito28. De esta manera, presentando estos antígenos, se generaría la selección negativa y posterior eliminación de los clones de LT autorreactivos. Sin embargo, ocurre que los LT específicos con baja afinidad son conservados. Estos LT autorreactivos pueden ser encontrados en la periferia en individuos sanos, y es probable que estén implicados en la patogénesis de los procesos autoinmunes del SNC. Es por ello que otros mecanismos mantendrían estos clones de LT autorreactivos bajo control en los órganos linfoides secundarios.
Tolerancia periférica y EM
a) T-regs en el modelo de encefalomielitis autoinmune experimental (EAE) en el ratón: evidencias indirectas
El modelo de EAE consiste en reproducir el fenómeno desmielinizante autoinmune mediante la inmunización con antígenos específicos de mielina en ratones. Pasado un breve lapso se genera una respuesta inmune contra los componentes de la mielina del SNC de los ratones a través de un mecanismo de reacción cruzada, originando las placas de desmielinización28. El desarrollo del proceso es similar a lo que ocurre en los seres humanos en la EM.
A lo largo del tiempo se observó que existía una población de LT comprometidos en la resolución del ataque inmunológico y la consecuente protección contra futuros ataques34. Esta población de LT no eran ni más ni menos que las T-regs. Luego de la identificación de esta población celular, se observó que la depleción de la misma en los ratones podía reiniciar el ataque inmunológico al infundir antígenos que en otras condiciones no podrían ser capaces de generarla34. Estos datos demostraron el papel central de las T-regs en el mantenimiento del umbral de tolerancia.
Una aproximación alternativa para evaluar la función de las T-regs ha sido incrementar el número de estas células al transferir células CD4+, CD25+ de ratones vírgenes a ratones en los cuales se había desarrollado la EAE35, 36. El experimento mostró que esa transferencia reducía la gravedad de la enfermedad en casi la totalidad de los ratones. El proceso por el cual se producía este fenómeno no ha sido completamente dilucidado, pero sería a través de los mecanismos previamente explicados de acción de las T-regs.
Otra de las observaciones realizadas con las T-regs en el modelo de EAE es la que demostró un aumento exponencial del número de T-regs durante el desarrollo del proceso, acumulándose estas células a nivel de las placas inflamatorias desmielinizantes en el SNC y contribuyendo posteriormente a la resolución de la enfermedad37, 38. Esta infiltración, estaba dada principalmente por parte de células CD4+ CD25+ Foxp3. Así mismo, la transferencia de los T-regs Foxp3 de ratones vírgenes potenciaba la recuperación de los ratones afectados no permitiendo la recurrencia de los fenómenos inflamatorios31, 39, 40.
De lo expresado previamente sobre las T-regs en el modelo de EAE se desprende que estas células tendrían una función central en la prevención de la agresión de los LT autorreactivos, además de mantener el umbral toleragénico alto frente a la autoagresión en condiciones pro-inflamatorias.
Esta capacidad de mantener el umbral de activación bajo condiciones inflamatorias significaría que la reacción cruzada inmunológica contra autoantígenos no sería capaz de desencadenar el proceso autoinmune inflamatorio, limitando de esta manera el riesgo de autoinmunidad41.
Las T-regs han sido también implicados en la recuperación una vez iniciado el proceso inflamatorio. El proceso de recuperación parecería estar mediado por la secreción activa de IL-10 y TGF-β38 por parte de estas células regulatorias.
Sin duda la función de las T-regs en la mediación y el control del modelo experimental de EAE serían cruciales.
b) T-regs en EM: evidencias directas
En los últimos años ha habido un incremento en el número de estudios dedicados a investigar la función de las T-regs en pacientes con EM5.
En los mismos, la frecuencia de T-regs en pacientes con EM no difería en comparación con controles sanos42- 44. Sin embargo varias líneas de investigación han demostrado que las T-regs en pacientes con EM se encuentran funcionalmente afectados o tienen una maduración defectuosa45, 46.
En estos experimentos, las T-regs de pacientes con EM fueron estimulados con mitógenos policlonales (Ac anti CD3 y anti CD28) y su capacidad supresora fue analizada mostrando una menor capacidad de supresión en evaluaciones in vitro. Estas T-regs defectuosas mostraron niveles más bajos de RNA mensajero de Foxp344, 46. Esta alteración de las T-regs sería la explicación por la cual la tolerancia periférica contra autoantígenos estaría desbalanceada, aumentando la susceptibilidad para el desarrollo de EM5. No estaría claro aún si esta disfunción tendría una relación causal en EM o representaría un defecto general dentro de la red regulatoria asociada con cualquier fenómeno autoinmune47.
Mas allá de las observaciones que relacionan la menor expresión de Foxp3 en las T-regs de pacientes con EM, el mecanismo exacto de disfunción es aún poco comprendido5.
La función de las T-regs en el control de la inflamación parenquimatosa en respuesta a autoantígenos es importante de remarcar. Las T-regs podrían combatir y destruir los componentes inflamatorios proveyendo así un mecanismo anti-inflamatorio protector en el SNC5. Estudios recientes en humanos -y concordante con lo observado en el modelo de EAE-, han demostrado un aumento del número de T-regs en el LCR en pacientes con EM comparados con la cantidad de T-regs en sangre periférica en estos mismos pacientes42. Este aumento en el LCR acompañado del descenso de las T-regs en sangre sugeriría que las T-regs serían reclutadas al sitio de inflamación42.
Recientemente se ha descrito una población de T-regs capaces de expresar HLA-G en humanos48. Estas células han demostrado ser hipoproliferativas, CD25- Foxp3- con propiedades supresoras potentes, mediada por el HLA-G. Se teoriza que estas células formarían parte de los mecanismos usados por el SNC para suprimir la inflamación y en contraste con las T-regs CD4+ CD25+ Foxp3+ de pacientes con EM no tendrían un deterioro en su función49, 50. Estas células T-regs HLA-G se acumularían en los sitios de inflamación frenando el proceso y evitando el ataque inmunológico50.
Interrelación entre CPA y T-regs en EM
Evidencias crecientes sugieren que las CPA son cruciales a través de su interacción directa sobre las T-regs y otros LT para inducir la respuesta inmune como la tolerancia ante antígenos específicos5.
Huang et al51 informaron la existencia de un elevado número de CPA en sangre periférica de pacientes con EM, así como en LCR. Estos datos soportan la teoría en la cual las CPA serían instrumentos primordiales en el mantenimiento y balance entre la actividad de los LT efectores y las T-regs en procesos inflamatorios del SNC.
Una evidencia pragmática de la interacción estaría dada por el efecto terapéutico beneficioso que tiene el interferón beta en pacientes con EM, esto se lograría al disminuir la expresión de moléculas de clase II en estas células e inducir y potenciar el efecto de las T-regs inducidas52, 53.
Más allá de lo presentado, varios aspectos permanecen actualmente especulativos en relación a la interacción de las CPA y las T-regs en el SNC.
T-regs como target inmunoterapéutico en EM
Varios obstáculos deberán ser resueltos antes que las T-regs puedan ser usadas terapéuticamente.
La primera limitación está dada por la especificidad antigénica de las T-regs. Aunque la transferencia policlonal de T-regs puede aliviar la gravedad de EAE54 esto no ha podido ser reproducido en otros laboratorios54.
Las T-regs antígeno específicas podrían controlar los procesos inmunes frente a desencadenantes específicos, siendo esto un problema en la EM donde el o los antígenos desencadenantes no se encuentran completamente identificados28.
Una segunda limitación en humanos es que el tratamiento debería frenar un proceso en marcha. Las T-regs precisamente no funcionan de manera óptima en un medio inflamatorio activo55. Como consecuencia, podrían ser necesarios los protocolos que combinan la proliferación de las T-regs e inactivan las LT autorreactivas para optimizar la eficacia de la intervención.
Sin duda, más allá de las limitaciones actuales, mucho esfuerzo en investigación se está llevando a cabo en el tema con el fin de encontrar una aplicación clínica a los hallazgos de laboratorio en el manejo de EM.
En conclusión, el estudio de las T-regs constituye un campo emergente en la práctica inmunológica de laboratorio así como en la práctica clínica.
Las T-regs son esenciales para el mantenimiento de la tolerancia inmunológica y su disfunción está asociada con el desarrollo de autoinmunidad órgano específica tanto en animales como en humanos.
Los datos disponibles actualmente sugieren que la disfunción temporaria o permanente de la función supresora de las T-regs estaría asociada con el desarrollo de enfermedades inflamatorias autoinmunes desmielinizantes del SNC. Si bien se ha avanzado en el conocimiento de los mecanismos por los cuales la falla en la función de esta población celular induciría al desarrollo de enfermedades autoinmunes, se desconoce el motivo de la falla, el origen y, fundamentalmente, cómo se podría prevenir la misma.
La traducción del conocimiento de estudios in vivo e in vitro hacia sus aplicaciones clínicas enfrenta una serie de retos; sin embargo, los estudios de investigación básica junto con la aplicación clínica serán esenciales para ampliar los conocimientos que poseemos en la actualidad sobre los mecanismos de tolerancia, autoinmunidad y fundamentalmente el rol de las T-regs en este fino balance inmunológico, así como sus posibles aplicaciones terapéuticas en enfermedades autoinmunes del SNC.
Agradecimiento: queremos agradecer a la Dra. María Virginia Paolini por su invaluable ayuda en la preparación del trabajo de revisión.
Conflictos de interés: Los autores no presentan conflictos de intereses relacionados con el trabajo de revisión.
1. Abbas B, Lichtman A, Pober J. Inmunología celular y molecular. Madrid, España: Interamericana. Mc Graw- Hill; 1998. [ Links ]
2. Patrucco L, Larriba J, Redal MA, Rojas JI, Argibay PF, Cristiano E. HLA-DRB1 and multiple sclerosis in Argentina. Eur J Neurol 2009;16: 427-9. [ Links ]
3. Cristiano E, Patrucco L, Rojas JI. A systematic review of the epidemiology of multiple sclerosis in South America. Eur J Neurol 2008;15: 1273-8. [ Links ]
4. Cristiano E, Patrucco L, Rojas JI, et al. Prevalence of multiple sclerosis in Buenos Aires, Argentina using the capture-recapture method. Eur J Neurol 2009;16: 183-7. [ Links ]
5. Zozulya AL, Wiendl H. The role of regulatory T cells in multiple sclerosis. Nat Clin Pract Neurol 2008;4: 384-98. [ Links ]
6. Astier AL, Hafler DA. Abnormal Tr1 differentiation in multiple sclerosis. J Neuroimmunol 2007; 191: 70-8. [ Links ]
7. O'Connor RA, Anderton SM. Foxp3+ regulatory T cells in the control of experimental CNS autoimmune disease. J Neuroimmunol 2008;193: 1-11. [ Links ]
8. Jiang H, Chess L. Regulation of immune responses by T cells. N Engl J Med 2006; 354: 1166-76. [ Links ]
9. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008;133: 775-87. [ Links ]
10. Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev 2006; 212: 28-50. [ Links ]
11. Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003; 198: 1875-86. [ Links ]
12. Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology 2006; 118:240-9. [ Links ]
13. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 2007; 8: 191-7. [ Links ]
14. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 2008; 8: 523-32. [ Links ]
15. Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med 2007;13: 108-16. [ Links ]
16. Annacker O, Pimenta-Araujo R, Burlen-Defranoux O, Barbosa TC, Cumano A, Bandeira A. CD25+ CD4+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. J Immunol 2001;166: 3008-18. [ Links ]
17. Ghiringhelli F, Menard C, Terme M, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med 2005; 202: 1075-85. [ Links ]
18. Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 2004; 104: 2840-8. [ Links ]
19. Zhao DM, Thornton AM, DiPaolo RJ, Shevach EM. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood 2006; 107: 3925-32. [ Links ]
20. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005; 22: 329-41. [ Links ]
21. Duthoit CT, Mekala DJ, Alli RS, Geiger TL. Uncoupling of IL-2 signaling from cell cycle progression in naive CD4+ T cells by regulatory CD4+CD25+ T lymphocytes. J Immunol 2005; 174: 155-63. [ Links ]
22. Oberle N, Eberhardt N, Falk CS, Krammer PH, Suri- Payer E. Rapid suppression of cytokine transcription in human CD4+CD25 T cells by CD4+Foxp3+ regulatory T cells: independence of IL-2 consumption, TGF-beta, and various inhibitors of TCR signaling. J Immunol 2007; 179: 3578-87. [ Links ]
23. Bluestone JA, Tang Q. How do CD4+CD25+ regulatory T cells control autoimmunity? Curr Opin Immunol 2005; 17: 638-42. [ Links ]
24. Tang Q, Adams JY, Tooley AJ, et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol 2006; 7: 83-92. [ Links ]
25. Tadokoro CE, Shakhar G, Shen S, et al. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med 2006; 203: 505-11. [ Links ]
26. Serrano Rios M, Moy CS, Martin Serrano R, et al. Incidence of type 1 (insulin-dependent) diabetes mellitus in subjects 0-14 years of age in the Comunidad of Madrid, Spain. Diabetologia 1990; 33: 422-4. [ Links ]
27. Fallarino F, Grohmann U, You S, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol 2006; 176: 6752-61. [ Links ]
28. Cassan C, Liblau RS. Immune tolerance and control of CNS autoimmunity: from animal models to MS patients. J Neurochem 2007; 100: 883-92. [ Links ]
29. Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res 1991; 28: 254-60. [ Links ]
30. Perry VH. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. J Neuroimmunol 1998; 90: 113-21. [ Links ]
31. Zhang X, Reddy J, Ochi H, Frenkel D, Kuchroo VK, Weiner HL. Recovery from experimental allergic encephalomyelitis is TGF-beta dependent and associated with increases in CD4+LAP+ and CD4+CD25+ T cells. Int Immunol 2006; 18: 495-503. [ Links ]
32. Kivisakk P, Mahad DJ, Callahan MK, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol 2004;55: 627-38. [ Links ]
33. Greter M, Heppner FL, Lemos MP, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 2005; 11: 328-34. [ Links ]
34. Anderton SM, Liblau RS. Regulatory T cells in the control of inflammatory demyelinating diseases of the central nervous system. Curr Opin Neurol 2008; 21: 248-54. [ Links ]
35. Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigenspecific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 2002;169: 4712-6. [ Links ]
36. Zhang X, Koldzic DN, Izikson L, et al. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol 2004; 16: 249-56. [ Links ]
37. McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol 2005; 175: 3025-32. [ Links ]
38. O'Connor RA, Malpass KH, Anderton SM. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J Immunol 2007; 179: 958-66. [ Links ]
39. Matsumoto Y, Sakuma H, Kohyama K, Park IK. Paralysis of CD4(+)CD25(+) regulatory T cell response in chronic autoimmune encephalomyelitis. J Neuroimmunol 2007;187: 44-54. [ Links ]
40. Gartner D, Hoff H, Gimsa U, Burmester GR, Brunner- Weinzierl MC. CD25 regulatory T cells determine secondary but not primary remission in EAE: impact on long-term disease progression. J Neuroimmunol 2006; 172: 73-84. [ Links ]
41. Anderton SM. Avoiding autoimmune disease-T cells know their limits. Trends Immunol 2006; 27: 208-14. [ Links ]
42. Feger U, Luther C, Poeschel S, Melms A, Tolosa E, Wiendl H. Increased frequency of CD4+ CD25+ regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin Exp Immunol 2007; 147: 412-8. [ Links ]
43. Haas J, Hug A, Viehover A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol 2005; 35: 3343-52. [ Links ]
44. Putheti P, Pettersson A, Soderstrom M, Link H, Huang YM. Circulating CD4+CD25+ T regulatory cells are not altered in multiple sclerosis and unaffected by disease-modulating drugs. J Clin Immunol 2004;24: 155-61. [ Links ]
45. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 2004;199: 971-9. [ Links ]
46. Hug A, Korporal M, Schroder I, et al. Thymic export function and T cell homeostasis in patients with relapsing remitting multiple sclerosis. J Immunol 2003;171: 432-7. [ Links ]
47. Baecher-Allan C, Hafler DA. Human regulatory T cells and their role in autoimmune disease. Immunol Rev 2006; 212: 203-16. [ Links ]
48. Feger U, Tolosa E, Huang YH, et al. HLA-G expression defines a novel regulatory T-cell subset present in human peripheral blood and sites of inflammation. Blood 2007;110: 568-77. [ Links ]
49. Wiendl H. HLA-G in the nervous system. Hum Immunol 2007;68: 286-93. [ Links ]
50. Wiendl H, Feger U, Mittelbronn M, et al. Expression of the immune-tolerogenic major histocompatibility molecule HLA-G in multiple sclerosis: implications for CNS immunity. Brain 2005;128: 2689-704. [ Links ]
51. Huan J, Culbertson N, Spencer L, et al. Decreased FOXP3 levels in multiple sclerosis patients. J Neurosci Res 2005; 81: 45-52. [ Links ]
52. Berghella AM, Totaro R, Pellegrini P, et al. Immunological study of IFNbeta-1a-treated and untreated multiple sclerosis patients: clarifying IFNbeta mechanisms and establishing specific dendritic cell immunotherapy. Neuroimmunomodulation 2005; 12: 29-44. [ Links ]
53. Trojano M, Pellegrini F, Fuiani A, et al. New natural history of interferon-beta-treated relapsing multiple sclerosis. Ann Neurol 2007; 61: 300-6. [ Links ]
54. Mekala DJ, Alli RS, Geiger TL. IL-10-dependent infectious tolerance after the treatment of experimental allergic encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Proc Natl Acad Sci U S A 2005; 102: 11817-22. [ Links ]
55. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003; 299: 1033-6. [ Links ]
Recibido: 29-6-2009
Aceptado: 14-10-2009

