










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680
Medicina (B. Aires) vol.70 no.2 Ciudad Autónoma de Buenos Aires Apr. 2010
ARTÍCULO ESPECIAL
Depresión y neuroplasticidad. Interacción de los sistemas nervioso, endocrino e inmune
Paola Cassano, Pablo Argibay
Instituto de Ciencias Básicas y Medicina Experimental, Instituto Universitario-Escuela de Medicina, Hospital Italiano de Buenos Aires, Argentina
Dirección Postal: Dra. Paola Cassano, Instituto de Ciencias Básicas y Medicina Experimental, Instituto Universitario-Escuela de Medicina, Hospital Italiano de Buenos Aires, Gascón 450, 1181 Buenos Aires, Argentina Fax: (54-11) 4954-0200 (int 5355) e-mail: paola.cassano@hospitalitaliano.org.ar
Resumen
La depresión clínica es una enfermedad física y psíquica que presenta bases neuropatológicas, sin embargo aún no se tiene un conocimiento exacto del origen o causas de esta enfermedad. Se conoce que existe un componente genético, aunque el componente ambiental en el desarrollo de la depresión es innegable. El estrés juega un rol esencial en el desencadenamiento de la depresión. La interacción y respuesta del sistema endocrino, inmune y nervioso se encuentran afectadas en este desorden. La observación de los efectos de los antidepresivos sobre la neurotransmisión monoaminérgica ha llevado hace muchos años a la hipótesis de las monoaminas de la depresión. Sin embargo, esta hipótesis ya no puede explicar muchos de los efectos de las drogas antidepresivas. La nueva hipótesis para explicar los efectos de los antidepresivos es la de neuroplasticidad neuronal. Esta hipótesis propone que los cambios que esas drogas producen sobre diversos sistemas, entre ellos el sistema nervioso, el inmune y el endocrino, son capaces de inducir cambios neuroadaptativos en el cerebro. La neuroplasticidad ha sido definida como la habilidad del cerebro para reorganizarse a sí mismo y formar nuevas conexiones neuronales a lo largo de la vida. Se propone que el mecanismo por el cual los antidepresivos logran sus efectos es mediante la neuroplasticidad.
Palabras clave: Depresión; Neuroplasticidad; Inflamación; Estrés
Abstract
Depression and neuroplasticity. Interaction of nervous, endocrine and immune systems. Clinical depression is a physical and psychic disease that has neuropathological basis, although the clear understanding of its ethiopathology is still missing. There is evidence of a genetic component in depression, however, the participation of environment is crucial. Stress plays an essential role in the onset of depression. The interaction and the response of the endocrine system with the immune and nervous system are altered in depression. The observation of the effect of antidepressants on monoaminergic transmitters leads to the hypothesis of monoamines. However this hypothesis cannot explain many of the mechanisms involved in the action of antidepressants. The new hypothesis proposed to explain the action of antidepressant is the neuroplasticity hypothesis. This hypothesis suggests that the effects of antidepressants on nervous, immune and endocrine systems are able to induce neuroadaptative changes in the brain. The neuroplasticity have been described as the ability of the brain to reorganize itself and form new neuronal connections throughout life. It is proposed that antidepressants influence neuroplasticity inducing improvements in the symptoms of this illness.
Key words: Depression; Neuroplasticity; Inflammation; Stress
La depresión mayor se presenta con características de humor depresivo, tristeza, pérdida de interés, falta de sensaciones de placer, conocido como anhedonia, sentimientos de culpa y poca autoestima, disturbios de sueño y apetito, poca energía y concentración pobre durante un período de 2 semanas según el Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR). La depresión mayor es una de las epidemias modernas que más compromete la calidad de vida. Los desórdenes asociados a ansiedad son también alarmantes, y muy frecuentemente están asociados a trastornos depresivos o abuso de sustancias psicotrópicas.
Estudios epidemiológicos demuestran que la depresión es un desorden hereditario1, 2. El polimorfismo en una región promotora del gen del transportador de serotonina es uno de los genes candidatos responsables de la enfermedad depresiva. La versión corta de este gen, llamado alelo s (por short en inglés) ha sido asociada con una baja eficiencia de transcripción del transportador de serotonina comparado con la versión larga, llamado l (por long en inglés). Frente a situaciones de mucho estrés, las personas s/l o s/s tienen mayor predisposición a sufrir depresión que las personas l/l1. Este estudio demuestra que un determinado genotipo, sólo es capaz de predisponer a la enfermedad pero siempre y cuando exista también el componente ambiental.
Es decir que la vulnerabilidad a sufrir depresión resulta genética sólo en cierta medida, existen factores no genéticos que son de gran importancia en el desencadenamiento de esta enfermedad. Entre ellos podemos mencionar factores como el estrés, traumas emocionales, infecciones virales, alteraciones que ocurren durante el desarrollo del cerebro, alteraciones neuroendocrinas, enfermedad vascular, Parkinson, daño cerebral, cierto tipo de cáncer, asma, diabetes y enfermedades autoinmunes, entre otras. De todos los factores mencionados, el estrés merece una atención especial, ya que muchas veces se describe a la depresión como un desorden relacionado al estrés, y no son pocos los casos en los que los episodios depresivos ocurren en un contexto de algún modo estresante2.
Tratamientos antidepresivos
Existen dos tipos fundamentales de tratamientos antidepresivos: la psicoterapia, cuyo principal instrumento es la palabra, y el abordaje clínico fisiológico, que incluye la intervención farmacológica principalmente.
El cerebro humano es increíblemente moldeable y las células pueden modificar su especialización después de un trauma, pueden "aprender" funciones nuevas (a este mecanismo se lo conoce como neuroplasticidad y lo ampliaremos más adelante). La comunicación neuronal parecería estar dañada en la depresión y los antidepresivos ayudan a restablecer los déficits funcionales y estructurales3. Muchas investigaciones han hipotetizado que la depresión podría aparecer por fallas del sistema nervioso en la plasticidad sináptica necesaria en respuesta al estrés. Esta falta de plasticidad sináptica es revertida por factores neurotróficos inducidos por el tratamiento con antidepresivos (ampliaremos los efectos de los antidepresivos sobre neuroplasticidad). Cuando se incrementan los niveles de serotonina, también se desarrollan funciones que corrigen el desequilibrio responsable del trastorno. Sin embargo, los mecanismos implicados en estos procesos no han sido todavía del todo dilucidados.
La hipótesis de las monoaminas
A raíz del descubrimiento de drogas capaces de revertir ciertos síntomas de la enfermedad depresiva, como los tricíclicos y los inhibidores de la mono-aminooxidasa (IMAO)4, se propuso que los sistemas en los que estas drogas actuaban (neurotransmisión serotoninérgica y norepinérfrica y en menor medida dopaminérgica) estarían involucrados entre las causas de la depresión5. Este descubrimiento llevó a la hipótesis de las monoaminas en la patofisiología de la depresión, la cual postulaba un déficit de serotonina y noradrenalina en áreas específicas de pacientes con depresión.
Luego, el entendimiento de los cambios químicos que ocurrían en el cerebro de personas o animales tratados con estas drogas, llevó a un mejor entendimiento de la función de las mismas y al descubrimiento de antidepresivos de segunda generación, más específicos en su función y por lo tanto con mejores resultados, como son los inhibidores específicos de la recaptación de serotonina y norepinefrina, tal como hemos descripto previamente.
Las mejoras en la transmisión monoaminérgica ocurren inmediatamente luego de la administración de los antidepresivos; sin embargo, tal como ampliaremos más adelante, los antidepresivos producen cambios en el humor luego de una administración prolongada, que puede variar de 3 semanas a unos pocos meses según el caso. Esto significaría que la mejora en la transmisión serotoninérgica o norepinérfrica no es la única responsable de las acciones clínicas de los antidepresivos. La hipótesis de las monoaminas ya no puede explicar del todo la efectividad de muchas drogas antidepresivas, como por ejemplo la tianeptina, cuya función es aumentar y no inhibir la recaptación de serotonina, y que resulta ser una efectiva droga antidepresiva5. También se ha demostrado6 que la depleción de serotonina o de norepinefrina no es capaz de inducir depresión clínica en sujetos sanos o de empeorar la sintomatología de pacientes con depresión mayor.
Lo mencionado anteriormente y mucha evidencia experimental han llevado a pensar en los antidepresivos no sólo como drogas que mejoran la neurotransmisión monoaminérgica sino también como drogas que inducen cambios neuroadaptativos, conocidos como neuroplasticidad en estructuras cerebrales afectadas por la ansiedad y la depresión. Y ha surgido de esta manera una nueva hipótesis conocida como hipótesis de plasticidad neuronal, que iremos detallando a continuación.
La neuroplasticidad
La neuroplasticidad es definida como la habilidad del cerebro para reorganizarse a sí mismo y formar nuevas conexiones neuronales a lo largo de la vida. Este fenómeno le permite a las neuronas del cerebro reparar daños cerebrales, adaptarse a enfermedades y ajustar sus actividades en respuesta a la nueva situación o cambios ambientales. La genética juega un papel muy importante en la plasticidad cerebral; sin embargo, el ambiente ejerce una pesada influencia en su mantenimiento. No debemos olvidar que la neuroplasticidad muchas veces contribuye al mal funcionamiento del sistema nervioso cuando la reorganización no resulta adecuada.
Numerosos procesos y sistemas son capaces de intervenir en la neuroplasticidad. Haremos una reseña de los mismos en relación a la enfermedad depresiva y finalmente explicaremos la hipótesis actual de plasticidad neuronal de los antidepresivos.
Psiconeuroendocrinoinmunología
La psiconeuroendocrinoinmunología concierne, tal como la propia palabra lo indica, a la interacción del comportamiento con los sistemas nervioso, endocrino e inmune. Esta interacción ocurre de manera bidireccional y ejerce una gran influencia en la salud y susceptibilidad a enfermedades. Las enfermedades físicas pueden desencadenar cambios psicológicos y viceversa. Se sabe que pacientes con cáncer sufren un estado depresivo durante un período de tiempo anterior a la aparición de su enfermedad, lo mismo ocurre con personas que padecen del virus HIV, que presentan alteraciones cognitivas y de memoria, aparte de cambios de comportamiento y del humor, antes de la aparición de los signos somáticos propios de la enfermedad. Hoy en día es muy aceptado que muchos de los cambios psicológicos y de comportamiento asociados con infecciones se deben a las citoquinas proinflamatorias producidas por la activación de células del sistema inmune tanto por infecciones como por estrés7.
Eje hipotálamo-pituitario-adrenal
Los glucocorticoides (principalmente cortisol en humanos y corticosterona en ratas) son hormonas esteroides que se encuentran involucradas en numerosos procesos fisiológicos, tanto en el cerebro como en el resto del organismo. Su secreción esta mediada por el eje hipotálamo pituitario adrenal (HPA), que es estimulado por acción del estrés a través de la hormona liberadora de corticotropina (CRF) en neuronas parvocelulares del núcleo paraventricular (PVN) del hipotálamo. CRF induce que la glándula pituitaria libere corticotropina a la sangre, que a la vez induce en la glándula adrenal la liberación de glucocorticoides. Los niveles de glucocorticoides se encuentran muy regulados mediante un mecanismo de retroalimentación inhibitoria, mediada por receptores de glucocorticoides tanto en el PVN como en la pituitaria.
La idea de que el estrés resulta perjudicial para un individuo surgió cuando se observó que una hiperactividad de la glándula adrenal y la pituitaria estaba asociada a alteraciones patológicas que podían ser revertidas cuando se normalizaba la actividad del eje HPA. Pero por otro lado, es importante recordar que la respuesta al estrés es beneficiosa desde el punto de vista que protege al individuo del daño y que la adaptación al mismo es una respuesta que debe aprenderse, ya que resulta esencial para futuras situaciones adversas. El "problema" surge cuando la situación de estrés se torna repetitiva y sostenida, en la cual la adaptación no resulta adecuada y aparecen cambios patológicos como consecuencia de los altos niveles de cortisol, alteraciones en el sistema inmune y alteraciones psicológicas. De esta manera, los desórdenes psicóticos, ansiedad y depresión, podrían surgir de este cambio en la actividad del eje HPA.
Tal como hemos descripto previamente, frente a una situación de estrés se libera la hormona CRF que estimula a la pituitaria para que libere ACTH y ésta a la vez a la glándula adrenal para que libere cortisol. La acción de CRF está mediada por los receptores CRFR1 y CRFR28. La activación del receptor tipo 1 en el PVN es responsable de la respuesta de huida o lucha, y en los corticotropos (células de la pituitaria anterior) de la liberación de ACTH. El receptor de tipo 2 sería el responsable de la respuesta adaptativa y recuperación a la situación de estrés9.
CRF también es capaz de activar al locus coeruleus, lo cual resulta en una activación del sistema simpático central que forma parte del sistema de respuesta al estrés agudo. Esta respuesta rápida es sensible al feedback negativo: el aumento agudo en los niveles de cortisol activa un tipo de receptores de cortisol (los mineralocorticoides, MR) en la pituitaria e hipotálamo y de esta manera disminuye la liberación de ACTH y consecuentemente de cortisol. Ante situaciones de alto estrés, o de estrés crónico, se activa otro tipo de receptores de cortisol (de los glucocorticoides, GR) en el cerebro, estos receptores son los que estarían alterados en pacientes con depresión. Individuos vulnerables al estrés parecen presentar un desequilibrio de estos receptores tal como revisan Revsin y Kloet en su trabajo10. Ampliamos este tema a continuación.
Receptores de glucocorticoides
Existen 2 receptores de glucocorticoides, el llamado receptor de glucocorticoides (GR por sus siglas en inglés glucocorticoid receptor) y el llamado receptor de mineralocorticoides (MR por sus siglas en inglés mineralocorticoid receptor)11. El MR tiene mayor afinidad y por lo tanto es ocupado frente a bajas concentraciones de glucocorticoides. El GR, al tener menor afinidad, requiere mayor concentración11, por lo cual se propone que el GR estaría involucrado en la respuesta de adaptación al estrés, cuando los niveles de glucocorticoides son mayores.
Los mayores niveles de GR y MR dentro del sistema nervioso central se encuentran en el hipocampo y en la pituitaria, ya por fuera de la barrera hematoencefálica12. El hipocampo presenta altas concentraciones de estos receptores. por lo cual, es una región sensible a la acción de los glucocorticoides. El mecanismo de acción de estos receptores se basa en el control de la tasa de transcripción de genes blancos mediante activación o represión transcripcional13-15. GR reprime su propia expresión mediante unión directa al ADN. Otro mecanismo de acción de los GR es lo que se conoce como cross talk transcripcional, que involucra la interacción con factores de transcripción o la competencia con estos por un mismo cofactor, como por ejemplo la interacción con NF-IB o proteínas CREB.
Aún no existe un consenso sobre el efecto de los glucocorticoides en la regulación de la expresión de GR y MR, algunos trabajos reportan que los mismos aumentan su expresión en presencia de glucocorticoides mientras que otros indican lo contrario16, 17. Esto mismo ocurre en cuanto a los estudios que relacionan el estrés con estos receptores, hay quienes hallaron que los incrementos fisiológicos de glucocorticoides debidos a estrés no serían suficientes para producir una down regulación de GR.
En cuanto a la relación de GR y depresión existe mucha evidencia que los vincula. Se propone que la función de GR está disminuida en pacientes con depresión mayor18. La corticosterona, o su análoga el cortisol en humanos, ha sido vinculada a síntomas depresivos, ya que el estrés juega un papel muy importante en el desencadenamiento de desórdenes afectivos. Una hiperactividad del eje HPA o mal funcionamiento de la sensibilidad del feedback mismo19, son las alteraciones neurobiológicas más importantes en la depresión mayor. Los pacientes depresivos presentan concentraciones altas de las hormona del eje HPA y exhiben respuesta exagerada de cortisol frente a la hormona adrenocorticotrópica (ACTH)20.
Debido a que la administración de CRF en animales de laboratorio lleva a cambios de comportamiento comparables con aquellos observados en la enfermedad depresiva, incluyendo alteraciones del humor, apetito, sueño, actividades locomotoras y de cognición21, se ha propuesto que la hipersecreción de CRH contribuiría a las características de comportamiento en la depresión mayor.
El mecanismo de resistencia a los glucocorticoides no se conoce en profundidad, pero se cree que sus receptores son los que estarían involucrados. La importancia de estos receptores GR y MR en los desórdenes depresivos aún no ha podido ser dilucidada, pero se propone que los efectos no se limitan sólo al feedback negativo de los glucocorticoides sino que también la activación o bloqueo de los mismos podrían modular sistemas asociados con memoria, respuestas de comportamiento, ansiedad y miedo 22,23. Por esta razón se propone que los GR y MR podrían estar mediando tanto los cambios de comportamiento y emocionales cómo así también las anormalidades en el feedback negativo en desórdenes depresivos.
El hipocampo ejerce un control indirecto muy importante sobre el eje HPA11, 24, y alteraciones del feedback debidas a daños hipocampales o pérdida celular conllevan a una pérdida de señalización que termina en una exposición acumulada a glucocorticoides que inducen una sobreexpresión patológica de esteroides en el cerebro e hipocampo. Existe una importante correlación entre los niveles plasmáticos de glucocorticoides y enfermedad con atrofia del hipocampo25, 26.
Aunque muchos investigadores creen que el número total de receptores de glucocorticoides parecería no influir en la depresión, otros sostienen que sí lo hacen27. Estudios recientes sugieren que la actividad del receptor, es decir su habilidad de unirse al ligando y translocar al núcleo, más que el número de receptores, sería el factor participante en los desórdenes del humor18. Esta falta de funcionalidad de GR podría ocurrir por mecanismos independientes a la unión al ligando, por ejemplo en la cascada de transducción de señales activada por moléculas no relacionadas con los corticosteroides, como por ejemplo IL1 y proteína quinasa A, las cuales por otro lado también han sido ampliamente relacionadas con la enfermedad depresiva (se detalla más adelante la participación del sistema inmune).
En pacientes con enfermedad depresiva que presentan disfunciones de estos receptores GR y MR28, el tratamiento con antidepresivos produce una mejora en el funcionamiento de estos receptores18, 29.
Estrés y serotonina
Se han observado evidencias que vinculan la concentración plasmática de cortisol con la serotonina. Entre ellas, se observó que la serotonina es capaz de activar a las neuronas del PVN para que liberen CRF30 y también de estimular la liberación de ACTH31. Por otro lado, el aumento agudo en los niveles de cortisol aumenta el turnover de serotonina debido a la estimulación de la actividad de la triptofano hidroxilasa32; sin embargo, en situaciones de estrés crónico el turnover de serotonina y su liberación disminuye. Esto estaría asociado a la activación de la enzima triptófano dioxigenasa33. También existen muchos trabajos realizados en roedores que demuestran una importante relación entre el estrés crónico y cambios en los sistemas serotoninérgicos que no expondremos en detalle, pero entre ellos se encuentran la inhibición del receptor 5HT1A, cambios en la afinidad del receptor 5HT2, cambios en la densidad del receptor 5HT1B, etc. Más allá de esta evidencia molecular obtenida en animales, en la clínica se sabe que la hipersecreción de cortisol resulta en una disminución de la concentración de triptofano (precursor de la serotonina).
La variante alélica s o l del gen del transportador de 5HT (tal como ha sido introducido previamente) podría estar relacionada a la susceptibilidad del paciente a los efectos del estrés.
Citoquinas en el SNC
Tanto la infección periférica como la inflamación llevan a la activación de macrófagos y de las células endoteliales con un aumento resultante de las citoquinas circulantes. Sin embargo, aparecen también ciertos síntomas gobernados por el SNC, por lo cual el cerebro de alguna manera se encuentra comunicado con el resto del organismo. Las citoquinas son moléculas grandes e hidrofílicas que no pueden atravesar la barrera hematoencefálica, por lo cual se ha propuesto que las citoquinas circulantes actúan directamente o indirectamente en el cerebro en sitios donde la barrera hematoencefálica se encuentra incompleta34, como en el OVLT, o que las citoquinas periféricas, vía activación de fibras aferentes que se proyectan en el cerebro, inducen la síntesis de citoquinas centrales que actúan directamente en el cerebro vía sus propios receptores.
La interleuquina 1 (IL1) circulante puede también actuar en neuronas del organum vasculosum de la lamina terminalis (OVLT) que contienen COX-2 (ciclooxigenasa 2) para inducir la secreción local de prostaglandinas, que difunden a otras áreas induciendo la liberación de IL1 dentro del cerebro35. La liberación de prostaglandinas del lado del cerebro de la barrera puede actuar como un mediador central de los efectos periféricos de las citoquinas. Se ha sugerido que las citoquinas circulantes pueden entrar en órganos circuventriculares e interactuar con células blanco que transducen la señal inmune en una señal secundaria en forma de prostaglandina que difunde libremente a otras células blanco vecinas.
Ciertas citoquinas como IL1 y IL6, y el factor tumoral de necrosis alfa (TNFa) aunque en principio fueron reconocidas por sus propiedades de señalización entre células inmunocompetentes, también son sintetizadas en el cerebro36. Existen receptores en el cerebro para estas moléculas que llevan a cambios importantes en la actividad neuronal. Las citoquinas tienen gran influencia en procesos del cerebro como actividad endocrina, sueño, comportamiento y neurodegeneración.
Estudios in vitro han demostrado que las células de la glía, astrocitos y microglía, expresan receptores para IL1, IL6 y TNFa; estos receptores también se encuentran en neuronas del hipocampo, hipotálamo y OVLT.
IL1 y IL6 causan la liberación del factor de liberación de CRF en el hipotálamo y en la pituitaria, la liberación de la hormona adrenocorticotrófica; lo cual da evidencia de que en el cerebro hay receptores para las dos citoquinas en cuestión37, 38.
Estrés y sistema inmune
La hipersecreción de glucocorticoides debido a situaciones de estrés crónico induce la activación de macrófagos tanto a nivel periférico como central, llevando a un aumento en los mediadores pro inflamatorios, principalmente incrementos de IL1b39. El estrés activa tanto al eje HPA como al sistema nervioso simpático, por lo cual afecta también al sistema inmune. Los linfocitos tienen receptores que responden a catecolaminas liberadas durante el estrés.
El glucocorticoide reduce la función de los linfocitos al actuar sobre los receptores de membrana de los mismos. La respuesta del sistema inmune al estrés está dada por un conjunto de señales que involucra catecolaminas, glucocorticoides, endorfinas y otros neuropéptidos. Los cambios que ocurren en el cerebro y en el sistema endocrino inevitablemente inducen cambios en las funciones inmunes, así también el sistema inmune es capaz de modular funciones de neurotransmisión central y endocrinas.
Las citoquinas proinflamatorias afectan las funciones monoaminérgicas centrales y también activan el eje HPA40, IL1, IL6 y TNF alfa; a su vez incrementan los cambios inflamatorios en el cerebro estimulando COX-2 para que produzca prostaglandina E2 que es capaz de modular la liberación de monoaminas en el cerebro41.
El estrés involucra estímulos que disturban el estado físico del organismo y resultan en una respuesta general y somática conocida como síndrome de adaptación general. El eje HPA juega un rol muy importante en la coordinación del sistema nervioso, endocrino e inmune.
La cascada neuroendocrina es disparada en neuronas vasopresinas y/o neuronas parvocelulares que expresan CRF del núcleo PVN, en células corticotrópicas de la pituitaria anterior que expresan ACTH y en células productoras de glucocorticoides de la corteza adrenal. La producción de glucocorticoides por la glándula adrenal produce una down regulación de la activación del eje HPA y afecta la producción periférica de citoquinas proinflamatorias a nivel de transcripción y de traducción. Estas citoquinas sobre-expresadas en el cerebro, a la vez conducen a una situación conocida como sickness behaviour que se describe a continuación.
También está demostrado que el eje HPA puede ser activado mediante la aplicación exógena de citoquinas como ser IL1, IL6 y TNFalfa42, 43.
Es decir, que existe una regulación bidireccional, el eje HPA es capaz de ejercer influencia sobre el sistema inmune y consecuentemente sobre moléculas proinflamatorias, y a la vez las citoquinas proinflamatorias son capaces de influenciar la liberación de hormonas relacionadas al eje HPA.
Además de sus efectos neuroquímicos, las citoquinas, como estresores, pueden influir en procesos importantes para la neuroplasticidad y así influir en el funcionamiento de procesos cognitivos y afectivos44. Estos efectos sobre la plasticidad podrían ser consecuencia de los efectos de las citoquinas sobre el eje HPA y viceversa, tal como hemos planteado anteriormente. En la Fig. 1 se esquematiza la interacción entre el sistema inmune, el sistema endocrino y el sistema nervioso.
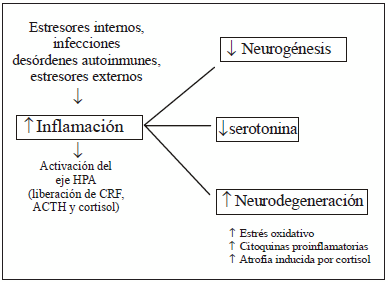
Fig. 1.- Participación del sistema inmune y endocrino en neuro-degeneración en la depresión.
Ambos sistemas, el inmune y el endocrino se encuentran alterados en la enfermedad depresiva; sin embargo, no se conoce aún el mecanismo que regula, o desregula, estos sistemas.
Neurogénesis y depresión
El rol de la neurogénesis en trastornos del ánimo como la depresión y desórdenes por estrés post traumáticos, se ha sugerido basado principalmente en estudios sobre el efecto del estrés y los tratamientos antidepresivos45.
Primates adultos sometidos a estrés psicosocial poseen menores niveles de proliferación en el hipocampo46, así mismo como ocurre en ratas expuestas a olores de sus depredadores47.
El tratamiento con antidepresivos aumenta la proliferación y la neurogénesis en hipocampo, y el tiempo en que esto ocurre coincide con los tiempo de acción terapéutica de los antidepresivos; este tiempo es de 2 a 3 semanas48. Por otro lado, se ha hipotetizado que la depression puede aparecer como consecuencia de una falla en la plasticidad sináptica en el SNC. Esta falla es revertida por factores neurotróficos que son inducidos por el tratamiento con antidepresivos, induciendo finalmente un aumento de la neurogénesis.
Hasta ahora había mucha evidencia de procesos de neurogénesis luego del tratamiento con antidepresivos en animales, pero recientemente se ha demostrado también en humanos49.
El estrés crónico produce atrofia en el hipocampo, y dicho efecto es revertido por la acción de los antidepresivos50 51. Sin embargo el dato más interesante que vincula las enfermedades mentales con características del hipocampo, es la evidencia de reducción de volumen hipocampal en pacientes con depresión mayor o desórdenes por estrés post traumático52, 53. Hay evidencia que indica que la reducción del volumen del hipocampo54 se correlaciona con el período de duración de la enfermedad depresiva, lo cual llevaría a pensar que esta disminución podría ser causada por la depresión 55.
Sin embargo, aún no se ha llegado a un consenso en cuanto a la relación entre el volumen hipocampal y este tipo de enfermedades. Esta reducción podría deberse a una disminución en el número de células granulares u otro tipo de células hipocampales o atrofia de neuronas hipocampales, producida por una disminución de la neurogénesis o atrofia de neuronas existentes.
Estudios utilizando un modelo de depresión conocido como paradigma de indefensión aprendida, demostraron una disminución en la proliferación de las células hipocampales y hallaron que el tratamiento con fluoxetina (antidepresivo Prozac®)) bloqueaba dicho efecto56. Otro trabajo que sustenta esta hipótesis, es uno en el que se demuestra que la neurogénesis sería necesaria para que la acción de algunos antidepresivos resulte efectiva57. Cabe destacar también que la inhibición de la neurogénesis no sería la principal causa de la enfermedad depresiva, pero sí estaría altamente involucrada en el proceso de reversión por antidepresivos.
A pesar de esta aparente reducción de la neurogénesis en modelos animales, las bases de esta reducción como sustrato de la depresión mayor aún son un tema en debate58.
Los conceptos actuales de neurobiología y de terapia para la depresión se focalizan en cambios neuroplásticos inducidos por estrés en circuitos neuronales específicos (como el sistema límbico y la corteza frontal)59, la reorganización o recuperación gradual de estas redes neuronales sería inducida por el tratamiento con antidepresivos60. La neurogénesis hipocampal ha sido el principal candidato involucrado en los efectos tardíos de los antidepresivos.
Hipótesis de plasticidad neuronal de los antidepresivos
Si bien la hipótesis de las monoaminas explicó en algún momento el mecanismo de acción de los antidepresivos, esta hipótesis es incapaz de explicar muchos otros efectos que tienen las drogas antidepresivas.
Existe evidencia de que el tratamiento con antidepresivos disminuye la síntesis y liberación de IL1, IL6 y TNF61, aumenta la síntesis de la citoquina antiinflamatoria IL10 y disminuye la de interferón gamma62. Se desconoce el mecanismo por el cual los antidepresivos producen alteraciones en la liberación de citoquinas, pero se propone que la elevación intracelular de la concentración de AMPc podría estar involucrada en la disminución de síntesis de citoquinas. En pacientes con esta enfermedad, se observó que el tratamiento con antidepresivos normalizaba los cambios que los mismos presentaban tanto a nivel de inmunidad celular como humoral63. También se ha propuesto que los antidepresivos mejorarían el funcionamiento del sistema inmune que posiblemente se encuentra afectado en pacientes depresivos, mediante un reestablecimiento del feedback del eje HPA, perdido en estos tipos de pacientes. Los antidepresivos actuarían produciendo una mejora de la sensibilidad de los receptores de glucocorticoides (GR y MR)64.
Otro mecanismo que ha sido también atribuido al efecto del tratamiento con antidepresivos, es la neurogénesis, proceso por el cual las células madres neuronales proliferan, se diferencian y sobreviven65. Si bien existe evidencia de que la neurogénesis es necesaria para que los antidepresivos resulten efectivos57, no se conoce con claridad si una disminución de la misma podría ser la causa, o una de las causas que desencadenarían la depresión. Hay estudios que revelan que pacientes con depresión poseen atrofia hipocampal52 (siendo el hipocampo la principal zona de neurogénesis en la adultez).
Factores de crecimiento, especialmente las neurotrofinas, juegan un rol esencial en la mediación de los efectos de los antidepresivos. El brain-derived neurotrophic factor (BDNF) se encuentra disminuido en pacientes depresivos y el tratamiento con antidepresivos es capaz de revertir esta situación66. Se ha estudiado también el efecto de los antidepresivos en la muerte celular programada y existe mucha evidencia que demuestra que el tratamiento con los mismos disminuye el número de células en apoptosis67.
Resumiendo, podemos decir que el tratamiento con antidepresivos induce adaptaciones en los numerosos sistemas involucrados en la enfermedad depresiva y produce una gran cantidad de cambios que en conjunto serían los responsables de las mejoras que producen. Entre ellos podemos mencionar la disponibilidad de neurotransmisores monoaminérgicos, la acción sobre los MR y GR, sus efectos como inmunomoduladores y la participación en procesos como la neurogénesis y la neurodegeneración. En la Fig. 2 se muestra la interacción de estos sistemas y la influencia de los antidepresivos en los mismos en relación a la depresión.

Fig. 2.- Factores y procesos involucrados en la depresión y el tratamiento con antidepresivos. (a) Existen varios factores que intervienen en el desencadenamiento de la enfermedad depresiva. Si bien existe un componente genético, el ambiente, principalmente el estrés y el eje HPA, juegan un rol esencial en este trastorno del ánimo. La depleción de catecolaminas, como así también procesos proinflamatorios y de neurodegeneración, que junto con una disminución de la neurogénesis llevan a una disminución del tamaño hipocampal son característicos de la depresión. (b) El tratamiento con antidepresivos es capaz de revertir la mayoría de las alteraciones encontradas en la enfermedad depresiva. No sólo aumenta los niveles de catecolaminas en el SNC, sino que también produce un aumento de la neurogénesis hipocampal que resulta esencial para la neuroplasticidad. Este tipo de drogas al mismo tiempo pueden regular negativamente la expresión de moléculas proinflamatorias y procesos de neurodegeneración, aparte de producir mejoras en el funcionamiento y regulación del eje HPA. Los antidepresivos producen cambios en el sistema nervioso que en conjunto conducen a una reorganización del mismo conocido como neuroplasticidad, capaz de revertir la sintomatología de la depresión.
La enfermedad depresiva es una enfermedad muy compleja, que como hemos descripto involucra la participación de numerosos sistemas, sistema nervioso, endocrino, inmune,y la participación del componente psicológico de quien la padece. De la misma manera, su tratamiento farmacológico involucra alteraciones en estos mismos sistemas, produciendo cambios neuroadaptativos que producen la mejora de los síntomas de la enfermedad.
Más estudios al respecto son necesarios para un mejor entendimiento de los mecanismos moleculares involucrados en la depresión. Muchas investigaciones sugieren que la neuroplasticidad podría ser clave en el desarrollo de nuevas drogas y tratamientos efectivos en relación a enfermedades psiquiátricas.
Conflictos de interés: Los autores no declaran conflictos de interés.
1. Caspi A, Sugden K, Moffitt TE, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003; 301: 386-9. [ Links ]
2. Wise MG, Taylor SE. Anxiety and mood disorders in medically ill patients. J Clin Psychiatry 1990; 51 Suppl: 27-32. [ Links ]
3. Castren E, Voikar V, Rantamaki T. Role of neurotrophic factors in depression. Curr Opin Pharmacol 2007; 7: 18-21. [ Links ]
4. Frazer A. Pharmacology of antidepressants. J Clin Psychopharmacol 1997; 17 Suppl 1: 2S-18S. [ Links ]
5. Hindmarch I. Beyond the monoamine hypothesis: mechanisms, molecules and methods. Eur Psychiatry 2002; 17 Suppl 3: 294-99. [ Links ]
6. Delgado PL, Moreno FA. Role of norepinephrine in depression. J Clin Psychiatry 2000; 61 Suppl 1: 5-12. [ Links ]
7. Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpe S, Maes M. IDO and interferon-alpha-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry 2005; 10: 538-44. [ Links ]
8. Reul JM, Holsboer F. Corticotropin-releasing factor receptors 1 and 2 in anxiety and depression. Curr Opin Pharmacol 2002; 2: 23-33. [ Links ]
9. de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci 2005; 6: 463-75. [ Links ]
10. Revsin Y, de Kloet ER. When glucocorticoids change from protective to harmful. Lessons from a type 1 diabetes animal model. Medicina (B Aires) 2009; 69: 353-8. [ Links ]
11. Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 1985; 117: 2505-11. [ Links ]
12. Reul JM, Pearce PT, Funder JW, Krozowski ZS. Type I and type II corticosteroid receptor gene expression in the rat: effect of adrenalectomy and dexamethasone administration. Mol Endocrinol 1989; 3: 1674-80. [ Links ]
13. Tronche F, Kellendonk C, Reichardt HM, Schutz G. Genetic dissection of glucocorticoid receptor function in mice. Curr Opin Genet Dev 1998; 8: 532-8. [ Links ]
14. Webster JC, Cidlowski JA. Mechanisms of Glucocorticoid- receptor-mediated Repression of Gene Expression. Trends Endocrinol Metab 1999; 10: 396-402. [ Links ]
15. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 2000; 21: 55-89. [ Links ]
16. Chao HM, Choo PH, McEwen BS. Glucocorticoid and mineralocorticoid receptor mRNA expression in rat brain. Neuroendocrinology 1989; 50: 365-71. [ Links ]
17. Herman JP. Spencer R. Regulation of hippocampal glucocorticoid receptor gene transcription and protein expression in vivo. J Neurosci 1998; 18: 7462-73. [ Links ]
18. Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry 2001; 49: 391-404. [ Links ]
19. Young EA, Haskett RF, Murphy-Weinberg V, Watson SJ, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry 1991; 48: 693-9. [ Links ]
20. Hermann B, Landgraf R, Keck ME, et al. Pharmacological characterisation of cortical gamma-aminobutyric acid type A (GABAA) receptors in two Wistar rat lines selectively bred for high and low anxiety-related behaviour. World J Biol Psychiatry 2000; 1: 137-43. [ Links ]
21. Nemeroff CB. The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry 1996; 1: 336-42. [ Links ]
22. De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev 1998; 19: 269-301. [ Links ]
23. Korte SM, de Boer SF, de Kloet ER, Bohus B. Anxiolyticlike effects of selective mineralocorticoid and glucocorticoid antagonists on fear-enhanced behavior in the elevated plus-maze. Psychoneuroendocrinology 1995; 20: 385-94. [ Links ]
24. Herman JP, Prewitt CM, Cullinan WE. Neuronal circuit regulation of the hypothalamo-pituitary-adrenocortical stress axis. Crit Rev Neurobiol 1996; 10: 371-94. [ Links ]
25. Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev 1986; 7: 284-301. [ Links ]
26. Landfield PW, Waymire JC and Lynch G. Hippocampal aging and adrenocorticoids: quantitative correlations. Science 1978; 202: 1098-1102. [ Links ]
27. Pariante CM, Nemeroff CB, Miller AH. Glucocorticoid receptors in depression. Isr J Med Sci 1995; 31: 705-12. [ Links ]
28. Heuser I, Deuschle M, Weber B, Stalla GK, Holsboer F. Increased activity of the hypothalamus-pituitary-adrenal system after treatment with the mineralocorticoid receptor antagonist spironolactone. Psychoneuroendocrinology 2000; 25: 513-8. [ Links ]
29. Pariante CM, Makoff A, Lovestone S, et al. Antidepressants enhance glucocorticoid receptor function in vitro by modulating the membrane steroid transporters. Br J Pharmacol 2001; 134: 1335-43. [ Links ]
30. Calogero AE, Bagdy G, Moncada ML, D'Agata R. Effect of selective serotonin agonists on basal, corticotrophin-releasing hormone- and vasopressin-induced ACTH release in vitro from rat pituitary cells. J Endocrinol 1993; 136: 381-7. [ Links ]
31. Owens MJ, Edwards E, Nemeroff CB. Effects of 5-HT1A receptor agonists on hypothalamo-pituitary-adrenal axis activity and corticotropin-releasing factor containing neurons in the rat brain. Eur J Pharmacol 1990; 190: 113-22. [ Links ]
32. Davis S, Heal DJ, Stanford SC. Long-lasting effects of an acute stress on the neurochemistry and function of 5-hydroxytryptaminergic neurones in the mouse brain. Psychopharmacology (Berl) 1995; 118: 267-72. [ Links ]
33. Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord 2007; 98: 143-51. [ Links ]
34. Hopkins SJ, Rothwell NJ. Cytokines and the nervous system. I: Expression and recognition. Trends Neurosci 1995; 18: 83-8. [ Links ]
35. Saper CB, Breder CD. Endogenous pyrogens in the CNS: role in the febrile response. Prog Brain Res 1992; 93: 419-28. [ Links ]
36. Schobitz B, De Kloet ER, Holsboer F. Gene expression and function of interleukin 1, interleukin 6 and tumor necrosis factor in the brain. Prog Neurobiol 1994; 44: 397-432. [ Links ]
37. Besedovsky H, Sorkin E. Network of immune-neuroendocrine interactions. Clin Exp Immunol 1977; 27: 1-12. [ Links ]
38. Besedovsky HO, del Rey A, Klusman I, Furukawa H, Monge Arditi G, Kabiersch A. Cytokines as modulators of the hypothalamus-pituitary-adrenal axis. J Steroid Biochem Mol Biol 1991; 40: 613-8. [ Links ]
39. Nguyen KT, Deak T, Will MJ, et al. Timecourse and corticosterone sensitivity of the brain, pituitary, and serum interleukin-1beta protein response to acute stress. Brain Res 2000; 859: 193-201. [ Links ]
40. Song C, Dinan T, Leonard BE. Changes in immunoglobulin, complement and acute phase protein levels in the depressed patients and normal controls. J Affect Disord 1994; 30: 283-8. [ Links ]
41. Connor TJ, Leonard BE. Depression, stress and immunological activation: the role of cytokines in depressive disorders. Life Sci 1998; 62: 583-606. [ Links ]
42. Himmerich H, Berthold-Losleben M, Pollmacher T. The relevance of the TNF-alpha system in psychiatric disorders. Fortschr Neurol Psychiatr 2009; 77: 334-45. [ Links ]
43. Silberstein S, Vogl AM, Bonfiglio JJ, et al. Immunology, signal transduction, and behavior in hypothalamic-pituitary- adrenal axis-related genetic mouse models. Ann N Y Acad Sci 2009; 1153: 120-30. [ Links ]
44. Seguin JA, Brennan J, Mangano E, Hayley S. Proinflammatory cytokines differentially influence adult hippocampal cell proliferation depending upon the route and chronicity of administration. Neuropsychiatr Dis Treat 2009; 5: 5-14. [ Links ]
45. Dranovsky A, Hen R. Hippocampal neurogenesis: regulation by stress and antidepressants. Biol Psychiatry 2006; 59: 1136-43. [ Links ]
46. Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci 1997; 17: 2492-8. [ Links ]
47. Tanapat P, Hastings NB, Rydel TA, Galea LA, Gould E. Exposure to fox odor inhibits cell proliferation in the hippocampus of adult rats via an adrenal hormone-dependent mechanism. J Comp Neurol 2001; 437: 496-504. [ Links ]
48. Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 2000; 20: 9104-10. [ Links ]
49. Boldrini M, Underwood MD, Hen R, et al. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009; 2376-89. [ Links ]
50. Magarinos AM, McEwen BS, Flugge G, Fuchs E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci 1996; 16: 3534-40. [ Links ]
51. Watanabe Y, Gould E, Daniels DC, Cameron H, McEwen BS. Tianeptine attenuates stress-induced morphological changes in the hippocampus. Eur J Pharmacol 1992; 222: 157-62. [ Links ]
52. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003; 160: 1516-8. [ Links ]
53. Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A 1996; 93: 3908-13. [ Links ]
54. Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry 2004; 161: 1957-66. [ Links ]
55. Sheline YI. Hippocampal atrophy in major depression: a result of depression-induced neurotoxicity? Mol Psychiatry 1996; 1: 298-9. [ Links ]
56. Malberg JE, Duman RS. Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology 2003; 28: 1562-71. [ Links ]
57. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003; 301: 805-9. [ Links ]
58. Kempermann G. Regulation of adult hippocampal neurogenesis - implications for novel theories of major depression. Bipolar Disord 2002; 4: 17-33. [ Links ]
59. Gass P, Henn FA. Is there a role for neurogenesis in depression? Biol Psychiatry 2009; 66: 3-4. [ Links ]
60. Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature 2008; 455: 894-902. [ Links ]
61. Xia Z, DePierre JW, Nassberger L. Tricyclic antidepressants inhibit IL-6, IL-1 beta and TNF-alpha release in human blood monocytes and IL-2 and interferon-gamma in T cells. Immunopharmacology 1996; 34: 27-37. [ Links ]
62. Kubera M, Kenis G, Bosmans E, Scharpe S, Maes M. Effects of serotonin and serotonergic agonists and antagonists on the production of interferon-gamma and interleukin- 10. Neuropsychopharmacology 2000; 23: 89-98. [ Links ]
63. Maes M, Smith R, Scharpe S. The monocyte-T-lymphocyte hypothesis of major depression. Psychoneuroendocrinology 1995; 20: 111-6. [ Links ]
64. Holsboer F, Barden N. Antidepressants and hypothalamic- pituitary-adrenocortical regulation. Endocr Rev 1996; 17: 187-205. [ Links ]
65. Schaffer DV, Gage FH. Neurogenesis and neuroadaptation. Neuromolecular Med 2004; 5: 1-9. [ Links ]
66. Lee BH, Kim H, Park SH, Kim YK. Decreased plasma BDNF level in depressive patients. J Affect Disord 2007; 101: 239-44. [ Links ]
67. Lee HJ, Kim JW, Yim SV, et al. Fluoxetine enhances cell proliferation and prevents apoptosis in dentate gyrus of maternally separated rats. Mol Psychiatry 2001; 6: 6, 725-8. [ Links ]
Recibido: 21-8-2009
Aceptado: 7-10-2009

