










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680
Medicina (B. Aires) vol.70 no.5 Ciudad Autónoma de Buenos Aires sep./oct. 2010
ARTÍCULO ESPECIAL
Molecular mechanisms of glucocorticoid receptor signaling
Marta Labeur, Florian Holsboer
Max Planck Institute of Psychiatry, Munich, Germany
Postal address: Dr. Marta Labeur, Max Planck Institute of Psychiatry, Kraepelinstrasse 10, 80804 Münich, Germany. Fax: (0049-89) 30622605 e-mail: labeur@mpipsykl.mpg.de
Abstract
This review highlights the most recent findings on the molecular mechanisms of the glucocorticoid receptor (GR). Most effects of glucocorticoids are mediated by the intracellular GR which is present in almost every tissue and controls transcriptional activation via direct and indirect mechanisms. Nevertheless the glucocorticoid responses are tissue -and gene- specific. GR associates selectively with corticosteroid ligands produced in the adrenal gland in response to changes of humoral homeostasis. Ligand interaction with GR promotes either GR binding to genomic glucocorticoid response elements, in turn modulating gene transcription, or interaction of GR monomers with other transcription factors activated by other signalling pathways leading to transrepression. The GR regulates a broad spectrum of physiological functions, including cell differentiation, metabolism and inflammatory responses. Thus, disruption or dysregulation of GR function will result in severe impairments in the maintenance of homeostasis and the control of adaptation to stress.
Key words: Glucocorticoid; Glucocorticoid receptor; Stress hormones
Resumen
Mecanismos moleculares de señalización del receptor de glucocorticoides. Esta revisión destaca los más recientes hallazgos sobre los mecanismos moleculares del receptor de glucocorticoides (GR). La mayoría de los efectos de los glucocorticoides son mediados por los GR intracelulares presentes en casi todos los tejidos y controlan la activación transcripcional por mecanismos directos e indirectos. Las respuestas a los glucocorticoides son específicas para cada gen y tejido. Los GR se asocian en forma selectiva con ligandos producidos en la glándula adrenal, corticosteroides, en respuesta a cambios neuroendocrinos. La interacción del ligando con el GR promueve: a) la unión del GR a elementos genómicos de respuesta a glucocorticoides, modulando la transcripción; b) la interacción de monómeros del GR con otros factores de transcripción activados por otras vías, llevando a la transrepresión. El GR regula un amplio espectro de funciones fisiológicas, incluyendo la diferenciación celular y las respuestas metabólicas e inflamatorias. Así, la desregulación de la función del GR resulta en graves defectos en el mantenimiento de la homeostasis y el control de la adaptación al estrés.
Palabras clave: Glucocorticoides; Receptor de glucocorticoides; Hormonas del estrés
In higher organisms, glucocorticoids synthesized by the adrenal cortex, play an important role in the adaptation to stressors and consecutive maintenance of internal homeostasis. The activation of the hypothalamic pituitary adrenal (HPA) axis by stressful situations -from emotional stress to infection-, leads to increase in plasma cortisol (in primates)/corticosterone (in rodents) concentrations, which in turn affect almost all physiological systems in the organism1. Besides the coordinated regulation of immune and neuronal responses, glucocorticoids regulate the endocrine system, in particular the HPA-axis, inhibiting corticotrophin releasing hormone (CRH), corticotrophin (ACTH) and consecutively their own synthesis that restores homeostasis. Glucocorticoids not only exert immunosuppressive and antiinflamatory actions, they also regulate cell-growth, bone-density, cardio-vascular function, metabolism, development and reproduction. Glucocorticoids have also an important impact in the brain on cognition, behaviour, mood and sleep2-4.
At the molecular level, the action of adrenal steroids is mediated by the glucocorticoid receptor (GR), a nuclear hormone receptor belonging to the superfamily of ligand-activated transcription factors. Three different 3'-splice variant of the GR have been shown: GR-α, the active form, GR -β, an inhibitor of GR-α function and GR-P, thought to be an activator of GR-α5. GR is predominantly localized within the cytoplasm as part of a complex with heat shock proteins and immunophilins. However, a continuous shuttling of the GR between the two cellular compartments takes place. Upon ligand binding, GR dissociates from the complex, where heat shock protein 90 (HSP90) plays a central role, and undergoes a conformational change6. Consecutively GR translocates to the nucleus. Activated GR induces or represses gene transcription either as homodimers or in a monomeric form. Glucocorticoids can also bind to mineralocorticoid receptors even with a higher affinity than the binding to GR. Moreover, when both receptors are coexpressed, they can form heterodimers that can bind to DNA with high affinity exerting singular properties7, 8.
Disregulation of the HPA axis that leads to excessive glucocorticoid secretion may have profound pathological effects in the organism. HPA dysfunction plays a critical role in the pathophysiology of mood disorders such as anxiety, depression and cognition impairment2.
GR gene variants have been associated to pathological conditions. Several polymorphisms have been described for GR. Among others, the N363S and BcII polymorphisms are associated with hypersensitivity to glucocorticoids and with increased abdominal fat mass, and major depression. In contrast the ER22/23EK polymorphism is related to glucocorticoid resistance and recent studies associate it with an increased risk of major depression9, 10. FKBP5, a co-chaperone of HSP90, regulates GR sensitivity. GR has less affitiny for glucocorticoids when FKBP5 is bound to the receptor complex. Polymorphisms in this gene are associated with increased expression of FKBP5 following GR activation, leading to a decreased negative feedback of glucocorticoids. This prolonged elevation of cortisol might be a risk factor for stress-related psychiatric disorders11. On the other hand certain polymorphisms in the FKBP5 gene may hasten the onset of antidepressant treatment response12. Moreover, hypersecretion of corticosteroids due to excessive adrenocorticotropic hormone (ACTH) secretion from a pituitary adenoma causes Cushing's disease, a severe clinical condition characterized by abnormal fat deposition around the neck, thinning of the skin, osteoporosis, insulin resistance, dyslipidemia, myopathy, amenorrhea, hypertension, anxiety and depression13, 14. These complex disorders still represents a major challenge for the physician in terms of efficient treatment. Understanding the molecular mechanisms of GR function may help in the development of new drugs for the treatment of Cushing's disease, stress-related disorders and other pathologies where excessive levels of corticosteroids play a causal role15.
In this review we will summarize recent findings related to the GR mediated molecular mechanisms of action (Fig. 1).
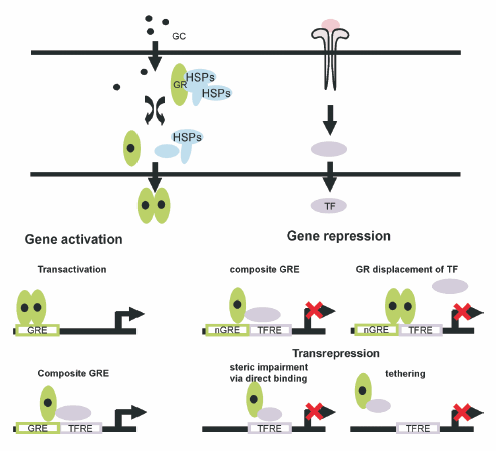
Fig. 1.- Transcriptional activities of GRs. Glucocorticoids can both activate and inhibit gene expression. Gene activation: GR homodimers bind to specific GRE in target genes and activate transcription. GR also contribute to gene activation by acting as coactivator for other TFs in composite GREs. Gene repression: several mechanisms of transcriptional repression have been documented for GR. By nGRE, GR displaces TFs or associates with them inhibiting gene transcription. By transrepression, GR inhibits the activity of TFs without DNA binding by GR (steric impairment or tethering). GC: glucocorticoids, GR: glucocorticoid receptor, GRE: glucocorticoid response element, nGRE: negative glucocorticoid response element, TF: transcription factor, TFRE: transcription factor response element, HSP: heat shock proteins. (The figure is presented in color in the web version of this article).
GR mediated transcriptional activation
Release of HSP90 allows GR translocation to the nucleus, homodimerization and DNA binding at glucocorticoid response elements (GREs) activating gene transcription (transactivation) in a cell and gene-specific manner16. GREs are palindromic sequences of two six base pair separated by a three base pairs spacer, present in the promoter of glucocorticoid-responsive genes. Deviation from this consensus sequence has been reported. Nevertheless GREs maintain important contacts with GR through specific functional groups on critical nucleotides within each palindromic sequence. Most of the genes mainly controlled by GR transactivation are involved in metabolic regulation; like increasing blood glucose levels, gluconeogenesis and mobilization of amino and fatty acids. When bound to DNA the glucocorticoid receptor interacts with transcriptional coactivator molecules, that stimulates transcription through direct interactions with the basal transcription machinery or by inducing local chromating remodelling, including histone acetylation or methylation, pointing to the roles of stress elicited glucocorticoids in mediating epigenetic modifications17-19. Some coactivators, like cyclic AMP response element binding protein (CBP) and p300 have histone acetyltransferase (HAT) activity and cause acetylation of core histones which facilitates the recruitment of RNA polymerase II and general transcription factors such as TATA box-binding protein (TBP)20, resulting in gene activation. Other coactivators recruit chromatin modifying enzymes to allow promoter activation, such as the members of the p160 family of coactivators, including steroid receptor coactivator (SRC)-1 (NcoA1), SRC-2 (TIF-2, GRIP1) and SRC-3 ( p/CIP, RAC3, ACTR or AIB1). These cofactors act as adaptor proteins linking the GR with other cofactors, like p300 and CBP, or with histone methyl transferases, like coactivator-associated arginine methyltransferase 1 and protein arginine methyltransferase 121.
Another common type of regulatory mechanism exerted by GR is their binding on composite GR-responsive regions, where additional transcription factors bind and efficiently induce glucocorticoid-mediated gene expression. Alternatively, the ligand-activated GR can modulate gene expression by interacting also with transcription factors, independently of binding to GREs. Transcription factors downstream to several signal transduction cascades, including cAMP response element-binding protein (CREB)22, activating protein 1 (AP-1)23, 24 and signal transducer and activator of transcription (STAT)25, 26 are described to interact with activated GR and promote transcription. Although crosstalk between NF-?B and GR leads to a mutual inhibition (see below), in some cases, activation of both transcription factors results in a cooperative environment27.
GR also recruits chromatin remodelling engines such as the mating-type switching/sucrose non-fermenting (SWI/SNF) complex. This complex is an ATP-dependent chromatin-remodeling factor with a multi-subunit structure28. It contains either BRG1 or hBrm as the central catalytic ATPase, as well as 10-12 BRG1-associated factors (BAFs). BRG1 alone can stimulate nucleosome remodelling, meanwhile addition of the core BAF subunits reconstitute chromatin remodelling to optimal levels29. SWI/SNF subunits can associate histone-modifying enzymes, transcription cofactors, tumor suppressor complexes and DNA replication factors. This ATP-dependent chromatin remodelling complex uses the energy derived from ATP hydrolysis to disrupt histone-DNA interactions in the context of GR activated transcription, thereby changing nucleosomal architecture leading to transcriptional activation. Although mainly related to transcriptional activation and considering that GRs can mediate either transcriptional activation or repression, it should be underlined that these enzymatic complexes have also been associated with transcriptional repression as demonstrated by activated corepressors30.
GR mediated gene repression
As mentioned, GR can suppress gene transcription by different mechanisms. Transrepression31, 32 accounts for many of the inhibitory effects glucocorticoids exert on the immune function and inflammatory processes33-35. In this mode of action, target genes are negatively regulated by GR via protein-protein interaction between GR and target transcription factors. This interaction may take place on promoters that do not contain GREs, with or without binding of the transcription factor to the DNA (steric impairment or tethering mechanism). Transrepression may take place on composite promoters as well, where GRE and transcription factors' responsive element may overlap. Thus, GR can interact with transcription factors like AP-136, NF-?B37 and STAT and inhibits their activity without involving direct GR binding to the DNA. Interaction between GR and AP-1 or NF-κB at the promoters of transcriptionally activated proinflammatory genes, accounts for many of the anti-inflammatory actions of glucocorticoids. Interestingly, the interaction between GR and STAT family members results also in regulation of the immune system, leading to a synergistic enhancement or inhibition of the transcriptional activity depending on the cellular context25. Recently, it has been found that GR interacts with the Th1-specific transcription factor T-Bet, thereby inhibiting T-Bet activity implicated in Th1 cell differentiation and inflammation38.
Corepressors, such as nuclear receptor corepressor (NcoR) and silencing mediator of retinoic acid and thyroid hormone receptor (SMRT), mediate gene repression in the absence of ligand or presence of antagonist by interacting with histone deacetylases (HDACs) and thereby compacting chromatin. Activated GR binds to coactivators to inhibit HAT activity directly and recruiting HDAC2, which reverses histone acetylation leading to suppression of activated inflammatory genes. Moreover, recently it was described that HDACs also deacetylate nonhistone proteins and HATs acetylates them. As other nuclear receptors, GR is acetylated upon binding of corticosteroid to GR. Consequently acetylated GRs translocate to the nucleus, bind GRE and activate genes. HDAC2 deacetylate the acetylated GR, which, for instance, enables GR to repress NF-kB activation of regulated inflammatory genes39.
Cofactors may change their function from being a coactivator to a corepressor. GRIP1 acts as a corepressor in GR-dependent regulation of AP-1 driven gene expression40. In the same direction, HDAC1 acts as a coactivator for the GR on the MMTV promoter41. Moreover, cofactor competition between GR and proinflammatory transcription factors have been described42.
Thus, depending on the gene, the transcription factor composition or cellular context, the gene transcription outcome may be of an inhibitory or stimulatory nature.
Alternatively GR can interact with negative GREs43 preventing transcription factors to bind to their binding site. Negative GREs are by far less frequent than positive GREs. At the hypothalamic and pituitary levels, corticotrophin-releasing hormone (CRH) and ACTH gene transcription are downregulated by negative GREs present in their promoters44, 45. However the molecular mechanism of GR mediated repression on these two gene targets seems to be different. Studies in GR mutant mouse models carrying a point mutation in the GR at the dimerization interface33, suggest that only the repression of pro-opiomelanocortin (POMC, gene coding for ACTH) in the anterior pituitary is dependent of GR dimerization. Monomeric GR still repress CRH gene expression, probably through protein-protein interaction with transcription factors present in that promoter, like AP-1, CREB and Nurr77. Thus, a monomeric form rather than GR homodimers would block the activity of DNA-bound transcription factors without contacting the DNA itself 46. Thus, even in the presence of negative GREs, GR can operate via different mechanisms, depending on the gene or tissue.
Inhibition of a target gene by GR may also alternatively take place at other regulatory points of signaling cascade. For instance, kinase-activating cascade, including MAPKs (p38, ERK, JNK) phosphorylates and activates inflammatory signals, like AP-1 or NF-?B target genes. However, all MAPKs have been identified as potential targets for activated GR through blockade of their activating phosphorylations. A novel aspect of GC transrepression has been shown by Beck et al 47. GR blocks NF-κB transcriptional activity, by upregulating IκBα, the cytoplasmatic NF-κB inhibitor48,49. In addition to the well described interaction-based mutual repression mechanism between the GR and NF-?B, additional mechanisms were recently discovered. It was shown the GR recruits the nuclear NF-κB kinase, mitogen- and stress-activated protein kinase-1 (MSK1) to the cytoplasm, thus modulating the chromatin environment and function of the inflammatory enhanceosoma. Loss of MSK1 at inflammatory gene promoter, causes inhibition of NF-kB transactivation47.
Modulation of GR by RNAs
More recently expanding numbers of noncoding RNAs (ncRNAs) with regulatory functions have been reported50. For example, growth arrest-specific 5 (Gas5), a single-strand ncRNA, was shown to interact with activated GR and suppressed GR-induced transcriptional activity of glucocorticoid-responsive genes by inhibiting binding of GR to target genes' GREs51. On the other hand, SRA (steroid receptor RNA activator) was reported to co-activate steroid receptors as an RNA transcript. It was shown that, even in the presence of cycloheximide, a protein synthesis inhibitor, transfected SRA enhanced endogenous GR activity in HeLa cells indicating that SRA acts as an RNA molecule rather than a protein52. These results have introduced a novel concept of regulation in nuclear receptor-mediated transcription and serve as another example of RNA transacted genomic information. Future studies will define more precisely the functional and physiological significance of RNA transcripts as components of protein complexes regulating transcription.
We conclude that the impact of glucocorticoids depends not only on the interplay among GR and DNA-regulatory elements, transcription factors, cofactors and chromatin remodelling complexes but also on the target gene and tissue. Moreover, signalling components of different signal transduction pathways originated from different stimuli, crosstalk, thereby increasing the level of complexity of gene regulation. Understanding the molecular mechanisms underlying glucocorticoids actions in the context of an integrated view of gene regulatory processes is necessary in order to design effective therapeutic strategies.
1. De Bosscher K, Vanden BW, Haegeman G. Mechanisms of anti-inflammatory action and of immunosuppression by glucocorticoids: negative interference of activated glucocorticoid receptor with transcription factors. J Neuroimmunol 2000; 109: 16-22. [ Links ]
2. Yu S, Holsboer F, Almeida OF. Neuronal actions of glucocorticoids: focus on depression. J Steroid Biochem Mol Biol 2008; 108: 300-9. [ Links ]
3. Muller MB, Uhr M, Holsboer F, Keck ME. Hypothalamic-pituitary-adrenocortical system and mood disorders: highlights from mutant mice. Neuroendocrinology 2004; 79: 1-12. [ Links ]
4. Muller MB, Keck ME, Zimmermann S, Holsboer F, Wurst W. Disruption of feeding behavior in CRH receptor 1-deficient mice is dependent on glucocorticoids. Neuroreport 2000; 11: 1963-6. [ Links ]
5. Russcher H, Dalm VA, de Jong FH, et al. Associations between promoter usage and alternative splicing of the glucocorticoid receptor gene. J Mol Endocrinol 2007; 38: 91-8. [ Links ]
6. Bledsoe RK, Montana VG, Stanley TB, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002; 110: 93-105. [ Links ]
7. Trapp T, Holsboer F. Heterodimerization between mineralocorticoid and glucocorticoid receptors increases the functional diversity of corticosteroid action. Trends Pharmacol Sci 1996; 17: 145-9. [ Links ]
8. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000; 23: 477-501. [ Links ]
9. van Rossum EF, Binder EB, Majer M, et al. Polymorphisms of the glucocorticoid receptor gene and major depression. Biol Psychiatry 2006; 59: 681-8. [ Links ]
10. Manenschijn L, van den Akker EL, Lamberts SW, van Rossum EF. Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Ann N Y Acad Sci 2009; 1179: 179-98. [ Links ]
11. Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009; 34 Suppl 1: S186-S195. [ Links ]
12. Binder EB, Salyakina D, Lichtner P, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nature Genet 2004; 36: 1319-25. [ Links ]
13. Labeur M, Theodoropoulou M, Sievers C, et al. New aspects in the diagnosis and treatment of Cushing disease. Front Horm Res 2006; 35: 169-78. [ Links ]
14. Labeur M, Arzt E, Stalla GK, Paez-Pereda M. New perspectives in the treatment of Cushing's syndrome. Curr Drug Targets Immune Endocr Metabol Disord 2004; 4: 335-42. [ Links ]
15. Revsin Y, de Kloet ER. When glucocorticoids change from protective to harmful. Lessons from a type 1 diabetes animal model. Medicina (Buenos Aires) 2009; 69: 353-8. [ Links ]
16. So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet 2007; 3: e94. [ Links ]
17. McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev 1999; 20: 321-44. [ Links ]
18. Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature 1996; 384: 641-3. [ Links ]
19. Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996; 87: 953-9. [ Links ]
20. Beato M, Sanchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev 1996; 17: 587-609. [ Links ]
21. Lonard DM, O'Malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell 2007; 27: 691-700. [ Links ]
22. Imai E, Miner JN, Mitchell JA, Yamamoto KR, Granner DK. Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J Biol Chem 1993; 268: 5353-6. [ Links ]
23. Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science 1990; 249: 1266-72. [ Links ]
24. Barrett TJ, Vig E, Vedeckis WV. Coordinate regulation of glucocorticoid receptor and c-jun gene expression is cell type-specific and exhibits differential hormonal sensitivity for down- and up-regulation. Biochemistry 1996; 35: 9746-53. [ Links ]
25. Liberman AC, Druker J, Perone MJ, Arzt E. Glucocorticoids in the regulation of transcription factors that control cytokine synthesis. Cytokine Growth Factor Rev 2007; 18: 45-56. [ Links ]
26. Stocklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature 1996; 383: 726-8. [ Links ]
27. Galon J, Franchimont D, Hiroi N, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J 2002; 16: 61-71. [ Links ]
28. Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: Molecular basis of biologic function. Steroids 2010; 75: 1-12. [ Links ]
29. Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell 1999; 3: 247-53. [ Links ]
30. Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003; 113: 207-19. [ Links ]
31. Newton R, Holden NS. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol 2007; 72: 799-809. [ Links ]
32. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids-new mechanisms for old drugs. N Engl J Med 2005; 353: 1711-23. [ Links ]
33. Reichardt HM, Kaestner KH, Tuckermann J, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998; 93: 531-41. [ Links ]
34. Reichardt HM, Tuckermann JP, Gottlicher M, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J 2001; 20: 7168-73. [ Links ]
35. Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 1997; 336: 1066-71. [ Links ]
36. Jonat C, Rahmsdorf HJ, Park KK, et al. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell 1990; 62: 1189-1204. [ Links ]
37. McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev 1999; 20: 435-59. [ Links ]
38. Liberman AC, Refojo D, Druker J, et al. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB J 2007; 21:1177-88. [ Links ]
39. Barnes PJ. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br J Pharmacol 2006; 148: 245-54. [ Links ]
40. Rogatsky I, Zarember KA, Yamamoto KR. Factor recruitment and TIF2/GRIP1 corepressor activity at a collagenase-3 response element that mediates regulation by phorbol esters and hormones. EMBO J 2001; 20: 6071-83. [ Links ]
41. Qiu Y, Zhao Y, Becker M, et al. HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Mol Cell 2006; 22: 669-79. [ Links ]
42. De BK, Vanden BW, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 2003; 24:488-522. [ Links ]
43. Cairns C, Cairns W, Okret S. Inhibition of gene expression by steroid hormone receptors via a negative glucocorticoid response element: evidence for the involvement of DNA-binding and agonistic effects of the antiglucocorticoid/antiprogestin RU486. DNA Cell Biol 1993; 12: 695-702. [ Links ]
44. Malkoski SP, Dorin RI. Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol Endocrinol 1999; 13:1629-44. [ Links ]
45. Drouin J, Sun YL, Nemer M. Glucocorticoid repression of pro-opiomelanocortin gene transcription. J Steroid Biochem 1989; 34: 63-9. [ Links ]
46. Dewint P, Gossye V, De BK, et al. A plant-derived ligand favoring monomeric glucocorticoid receptor conformation with impaired transactivation potential attenuates collagen-induced arthritis. J Immunol 2008; 180: 2608-15. [ Links ]
47. Beck IM, Vanden BW, Vermeulen L, et al. Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-kappaB inhibition. EMBO J 2008; 27: 1682-93. [ Links ]
48. Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS, Jr. Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science 1995; 270: 283-6. [ Links ]
49. Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 1995; 270: 286-90. [ Links ]
50. Mattick JS. The functional genomics of noncoding RNA. Science 2005; 309: 1527-8. [ Links ]
51. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal 2010; 3: ra8. [ Links ]
52. Lanz RB, McKenna NJ, Onate SA, et al. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 1999; 97: 17-27. [ Links ]
Recibido: 2-5-2010
Aceptado: 3-6-2010

