










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680
Medicina (B. Aires) vol.70 no.6 Ciudad Autónoma de Buenos Aires nov./dic. 2010
ARTÍCULO ORIGINAL
Molecular characterization of severe and mild cases of influenza A (H1N1) 2009 strain from Argentina
Elsa Baumeister1, Gustavo Palacios2, Daniel Cisterna1, Alexander Solovyov2, 3, Jeffrey Hui2, Nazir Savji2, Ana Valeria Bussetti2, Ana Campos1, Andrea Pontoriero1, Omar J. Jabado2, Craig Street2, David L. Hirschberg2, Raul Rabadan3, Virginia Alonio1, Viviana Molina1, Stephen Hutchison4, Michael Egholm4, W. Ian Lipkin2
1Instituto Nacional de Enfermedades Infecciosas, ANLIS Dr. Carlos G. Malbrán, Buenos Aires, Argentina,
2Center for Infection and Immunity, Mailman School of Public Health, Columbia University, New York, New York, USA,
3Department of Biomedical Informatics, Center for Computational Biology and Bioinformatics, Columbia University, New York, New York, USA,
4454 Life Sciences, Branford, Connecticut, USA
Postal address: Dra. Elsa Baumeister, Instituto Nacional de Enfermedades Infecciosas, ANLIS Dr. Carlos G. Malbrán, Av. Vélez Sarsfield 563, 1281 Buenos Aires, Argentina
Fax: (54-11) 4301-1035 e-mail: ebaumeister@anlis.gov.ar
Abstract
While worldwide pandemic influenza A(H1N1) pdm case fatality rate (CFR) was 0.4%, Argentina's was 4.5%. A total of 34 strains from mild and severe cases were analyzed. A full genome sequencing was carried out on 26 of these, and a partial sequencing on the remaining eight. We observed no evidence that the high CFR can be attributed to direct virus changes. No evidence of re-assortment, mutations associated with resistance to antiviral drugs, or genetic drift that might contribute to virulence was observed. Although the mutation D225G associated with severity in the latest reports from the Ukraine and Norway is not observed among the Argentine strains, an amino acid change in the area (S206T) surrounding the HA receptor binding domain was observed, the same previously established worldwide.
Key words: H1N1 pdm; Influenza; Argentina
Resumen
Caracterización molecular de cepas de influenza A (H1N1) 2009 de casos leves y graves de la Argentina. Mientras que la tasa de letalidad (CFR) para (H1N1)pdm en todo el mundo era del 0.4%, en la Argentina la mortalidad observada fue de 4.5%. La secuenciación del genoma completo de 26 cepas de virus argentinos de influenza A (H1N1)pdm de casos leves y graves y de 8 cepas secuenciadas parcialmente no mostró evidencia de que la elevada tasa de letalidad se pueda atribuir directamente a cambios en el virus. No se encontraron hallazgos de recombinación, de mutaciones asociadas con la resistencia a los medicamentos antivirales ni de variaciones genéticas que puedan contribuir a la virulencia observada. Si bien la mutación D225G asociada con la gravedad, comunicada en informes procedentes de Ucrania y Noruega, no se ha encontrado en las cepas argentinas estudiadas, se ha observado un cambio aminoacídico en la región (S206T) en torno al dominio del sitio de unión al receptor en la HA, el mismo hallado en cepas distribuidas alrededor del mundo.
Palabras clave: H1N1 pdm; Influenza; Argentina
On June 11, 2009, in acknowledgement of sustained global human to human transmission, the WHO declared that the novel influenza A virus (H1N1pdm) was pandemic. Although H1N1pdm has not been reported as more virulent than seasonal H1N1 influenza, its high transmissibility in an immunologically naïve population implies potential for substantial morbidity and mortality and mandates close surveillance for evolution toward increased virulence. Based on worldwide surveillance data and mathematical modeling, the case fatality rate (CFR) of H1N1pdm was estimated at 0.4%1, 2. In contrast, although the first case in Argentina was detected in May 17, 3056 cases with 137 deaths were reported by July 16, representing a computed CFR of 4.5%2.
The pathogenesis of infectious diseases is the result of the interaction between the pathogen, the host and the environment (e.g. secondary infections, health care system). Although it is likely that severity will vary from country to country depending on health care resources and the public health measures adopted to mitigate impact, a second possible cause for a significantly higher mortality rate may be changes in the virus itself. Influenza viruses may change in virulence and host range as a function of reassortment with strains that contain virulence factors3, or through genetic drift. To investigate these possibilities as explanations for the increased CFR in Argentina we sequenced the genomes of H1N1pdm virus obtained from cases of mild and severe disease.
Materials and Methods
Samples collected included: 50 nasal swabs containing H1N1pdm and 50 virus isolates. Severe cases were defined as having a fatal outcome (n = 21) or requiring ventilation support (n = 15), (Table 1). Samples here analyzed had been routinely collected for use in approved H1N1 tests at INEI-ANLIS Malbrán. Samples were identified before being re-analyzed at Columbia University. Data were analyzed anonymously.
TABLE 1.- Data for the specimens presented in this study

*NS: nasal swab
RNA was isolated using TriReagent (Molecular Research Center, Cincinnati, OH, USA). DNA was removed from RNA preparations by treatment with DNase I (DNA-free, Ambion, Austin, TX, USA). Reverse transcription (RT) reactions were performed using the Superscript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA).
Two protocols were used to amplify template for 454 pyrosequencing (Roche Life Sciences, CT, USA). In one protocol, first strand synthesis was initiated with a random octamer linked to an specific artificial primer sequence known as the sequence independent amplification (SIA) primer4; a modified protocol was also used wherein first strand synthesis was initiated with the SIA primer doped with a primer mixture containing the same specific sequence linked to influenza virus A (FLUAV), influenza virus B (FLUBV) and influenza virus C (FLUCV) sequences representing the conserved termini of influenza virus genome segments5. Random libraries prepared with this method were pyrosequenced. The raw sequencing reads were trimmed and analyzed using GS Reference Mapper (Roche Life Sciences, CT, USA) against the reference sequence A/H1N1/4/California/2009 (accession numbers FJ966079- FJ966086).
Alternatively, random primed cDNA were amplified with primers spanning the whole genome of H1N1pdm for conventional Sanger sequencing. Primers were designed using SCPrimer (http://scprimer.cpmc.columbia.edu)6 to amplify 800 bp regions of the H1N1pdm genome. A 500 bp staggered overlap strategy was employed to improve fidelity of the sequences. Terminal sequences were added by PCR using a universal influenza A primer, targeting the conserved viral termini (5' ATA TCG TCT CGT ATT AGT AGA AAC AAG G,)7 combined with specific primers positioned near the ends of the genome segments. Real-time PCR SYBR Green assays (Applied Biosystems, Foster City, CA) were conducted to determine the viral load in each sample (primers available upon request). The libraries yielded between 2475 and 42 188 non-host sequence reads. Mapping indicated coverage ranging from 21% to 99% distributed along the influenza virus 8 segments. Virus sequence representation was directly proportional to viral load. A titer of at least 20 000 genome copies/reaction was necessary to achieve >90% coverage.
For the clustering analysis we performed the Principal Component Analysis (PCA) followed by consensus K-means clustering. A consensus clustering out of the 1000 individual K-means trials was built. Parsimony tree was built of the 8 concatenated segments using the dnapars program from the PHYLIP package8. Clusters previously found with the K-means algorithm correspond to particular clades in the tree.
Results
Twenty-six complete genomes and eigth partial genomes containing complete HA and NA gene were obtained (Table 1). They were analyzed for re-assortment, presence of mutations that confer antiviral resistance and phylogenetic clustering. All 26 genomes obtained contained the S31N mutation in the M2 protein, which confers cross-resistance to the adamantine class of anti-influenza drugs. The most commonly detected mutation in oseltamivir-resistant viruses (H274Y) was not detected in any of the genomes. None of the Argentinean genomes contain the mutation D225G in within the influenza HA receptor binding site (Fig. 1). No re-assortment with regular seasonal influenza strains (either H1N1 or H3N2) was detected.
Fig. 1.- Amino acid alignment (in one-letter code) within the influenza HA receptor binding site (position 171- 237; H3 numbering). Positions that had been implicated in receptor specificity are indicated by ^. Positions 183, 190, 222, 225, 226, 2279-11, 13 and 189, 193 and 1959 are marked. Positions with amino acid sequence changes are highlighted. The box over position 206 highlights the amino acid change in Cluster 5 HA segments. Original host and type of receptor binding specificity are also indicated; Swine (H) are human isolates of influenza virus of swine origin.
By analysis of the eigenvectors of PCA, we selected 17 single nucleotide polymorphims (SNPs) that reliably classified 5 clusters, wherein the difference between any pair of clusters was at least 2 SNPs (Table 2). Each of the clusters is named after the earliest isolate that belongs to it. Our analysis identifies five different clusters of H1N1pdm sequences. Cluster 1 is composed of sequences first identified in Mexico in early April and sequences obtained shortly thereafter in the US, Canada, China and Europe. Cluster 2 contains sequences detected in California, US in early April and their relatives, isolated in the US, Mexico, Canada, Dominican Republic, Japan, China and Europe. Cluster 3 is defined by sequences obtained in Japan in the second half of May. Cluster 4 is composed of isolates from New England, US and Canada obtained in May and June. Cluster 5 is the largest cluster, dates back to the earliest isolates from Mexico (April 19, 2009) and has wide geographical spread, including Mexico, the United States, Argentina, Canada, China and several European countries. Specimens from Argentina belong to cluster 5. It is distinguished by 6 mutations: G2163A(PB2) G658A(HA) G1248A(NP) G492A(MA) G600A(MA) A367G(NS) (Fig.2). The Argentinean isolates (except for #8989) contain a characteristic polymorphic marker T1623C in the PA segment. This substitution does not change the amino acid.
TABLE 2.- Single Nucleotide Polymorphisms (SNPs) used to classify 5 clusters
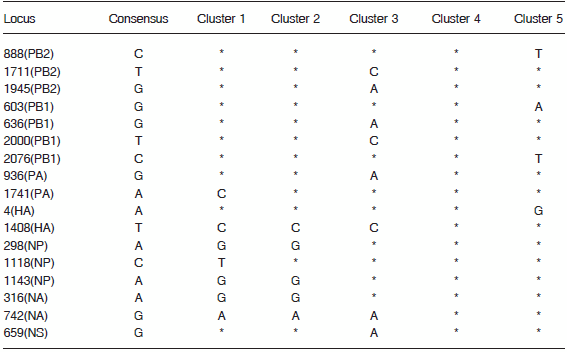
*: no change compared with the consensus
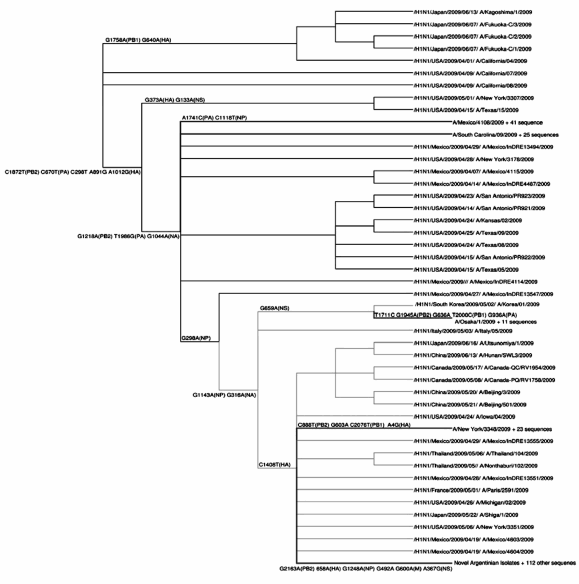
Fig. 2.- Phylogenetic analysis of the H1N1pdm sequences using the Maximum Parsimony method. For building the tree we use the PHYLIP package8, and for visualizing it we use the Dendroscope program17.
Discussion
PCA analyses revealed that all Argentinean isolates are related suggesting a recent common origin, but given that the newer splits (especially those in the Cluster 5) cannot be resolved completely, we cannot determine whether there was a single H1N1pdm introduction in Argentina or several events involving highly related viruses. Interestingly, one of the phylogenetically informative sites that define a large sub-clade in cluster 5 is the transversion A-T in position 658, that results in a Thr-Ser change in position 206 (H3 numbering). This change occurs in the defined receptor-binding site of FLUAV H1N1. Since the change is observed in both clinical samples and virus isolates, it cannot be attributed to selection in cell culture. It has been modeled extensively and the sites of interaction between the sialic based carbohydrates and the hemagglutinin are well characterized9-13. Moreover, this same region had been described as one of the more significant antigenic epitopes of H1-subtype influenza A virus and related with vaccine efficacy14. Three secondary structure elements, the 190 helix (residues 190-198); the 130 loop (residues 135 to 138), and the 220 loop (residues 221 to 228) form the sides of the receptor-binding subdomain. Changes in this area (D225G, D190E, K222L9, 11) or others outside of this area that result in a change in the conformation of the domain (e.g. Thr18913), result in changes in the affinity of the receptor-binding site and are believed to be responsible for human, swine or avian hosts adaptation. In addition, the substitution K222L results in complete abolition of binding9. Of note, the mutation D225G in this same area has also been observed in fatal cases from the Ukraine and Norway15, 16.Given the limited availability of data on the H1N1pdm strains, we have not been able to demonstrate if this change is positively selected.
In summary, sequencing results of 26 complete genomes of H1N1pdm virus obtained from cases of mild and severe disease indicate continuous virus evolution by drift, but show no evidence that the Argentinean high case fatality rate can be attributed to changes in viral virulence or resistance to antivirals.
Acknowledgements: We would like to thank all the personnel of the INEI-ANLIS Dr. Carlos G. Malbrán. This work would not have been possible without their help during the peak of the outbreak. Especially, we would like to thank all the personnel of the Virology Department, where the majority of the sample handling and testing was performed. This work was supported by the National Institutes of Health awards AI051292, U54 AI57158 (Northeast Biodefense Center), NO1 AI30027, Google.org, the United States Department of Defense and the Ministry of Science, Technology and Productive Innovation of Argentina, ANLIS Institutional Strengthening Plan.
Conflicts of interest: The funding organizations had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. S. Hutchison and M. Egholm are 454 Life Sciences employees.
1. Fraser C, Donnelly CA, Cauchemez S, et al. Pandemic potential of a strain of influenza A (H1N1): early findings. Science 2009; 324: 1557-61. [ Links ]
2. Vaillant L, La Ruche G, Tarantola A, Barboza P. Epidemiology of fatal cases associated with pandemic H1N1 influenza 2009. Euro Surveill 2009; 14(33), pii: 19309 [ Links ]
3. Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol 2006; 80: 7976-83. [ Links ]
4. Palacios G, Quan PL, Jabado OJ, et al. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg Infect Dis 2007; 13: 73-81. [ Links ]
5. Quan PL, Palacios G, Jabado OJ, et al. Detection of respiratory viruses and subtype identification of influenza A viruses by GreeneChipResp oligonucleotide microarray. J Clin Microbiol 2007; 45: 2359-64. [ Links ]
6. Jabado OJ, Palacios G, Kapoor V, et al. Greene SCPrimer: a rapid comprehensive tool for designing degenerate primers from multiple sequence alignments. Nucleic Acids Res 2006; 34: 6605-11. [ Links ]
7. Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 2001; 146: 2275-89. [ Links ]
8. Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.69. Distributed by author; 2009. [ Links ]
9. Stevens J, Blixt O, Glaser L, et al. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol Biol 2006; 355: 1143-55. [ Links ]
10. Stevens J, Corper AL, Basler CF, Taubenberger JK, Palese P, Wilson IA. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 2004; 303: 1866-70. [ Links ]
11. Tumpey TM, Maines TR, Van Hoeven N, et al. A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science 2007; 315: 655-9. [ Links ]
12. Childs RA, Palma AS, Wharton S, et al. Receptor-binding specificity of pandemic influenza A (H1N1) 2009 virus determined by carbohydrate microarray. Nat Biotechnol 2009; 27: 797-9. [ Links ]
13. Gamblin SJ, Haire LF, Russell RJ, et al. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 2004; 303: 1838-42. [ Links ]
14. Deem MW, Pan K. The epitope regions of H1-subtype influenza A, with application to vaccine efficacy. Protein Eng Des Sel 2009; 22: 543-6. [ Links ]
15. Shen J, Ma J, Wang Q. Evolutionary trends of A(H1N1) influenza virus hemagglutinin since 1918. PLoS One 2009; 4(11): e7789. [ Links ]
16. ProMED-mail. Mutation of pandemic influenza A (H1N1) 2009 virus in Norway. ProMED-mail 2009, Fri 20 Nov 2009. [ Links ]
17. Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics 2007; 8: 460. [ Links ]
Recibido: 26-1-2010
Aceptado: 27-9-2010

