










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680
Medicina (B. Aires) vol.71 no.4 Ciudad Autónoma de Buenos Aires jul./ago. 2011
ARTÍCULO ESPECIAL
Epigenética y epigenoma: Un paso más allá en la etiología y potencial tratamiento de las enfermedades neurológicas
Sergio J. González1, Edgardo Cristiano2, Pablo Argibay1
1Unidad de Medicina Molecular y Genómica, Instituto de Ciencias Básicas y Medicina Experimental (ICBME),
2Servicio de Neurología, Hospital Italiano de Buenos Aires
Dirección postal: Dr. Pablo Argibay, Unidad de Medicina Molecular y Genómica, Instituto de Ciencias Básicas y Medicina Experimental, Hospital Italiano de Buenos Aires, Gascón 450, 1181 Buenos Aires, Argentina
Fax (54-11) 4959-0200 (int. 8919) e-mail: pablo.argibay@hospitalitaliano.org.ar
Resumen
Los mecanismos de regulación epigenética poseen una función muy importante en el desarrollo y en la función de todos los sistemas del organismo. El fracaso en el normal mantenimiento de esta regulación y desarrollo, debido a factores ambientales, podría contribuir al desarrollo de múltiples enfermedades en pacientes predispuestos genéticamente. Si bien la fisiopatología de gran parte de las enfermedades es desconocida en sus mecanismos moleculares más finos, existen evidencias de que la mayoría de los mecanismos fisiopatológicos convergerían a una interacción entre los genes y el ambiente. La epigenética nos permitiría estudiar la interacción mencionada en los niveles moleculares de regulación de la expresión o supresión de un gen y su relación con diversas enfermedades. Particularmente las enfermedades neurológicas, por su complejidad, la gran cantidad de genes involucrados y la influencia ambiental, son un ámbito más que apropiado para investigar potenciales mecanismos epigenéticos en su fisiopatogenia y desarrollo. La presente revisión tiene como objetivo describir en detalle los mecanismos de regulación epigenética y los hallazgos conocidos que provocarían la disfunción de estos mecanismos como posibles desencadenantes de diferentes enfermedades neurológicas.
Palabras clave: Epigenética; Expresión génica; Ambiente
Abstract
Epigenetics and epigenome: A step forward in the etiology and potential treatment of neurological diseases. The mechanisms of epigenetic regulation play an important role in the development and function of body systems. Failure in the maintenance of this regulation as well as environmental factors could contribute to the development of multiple disease s in genetically predisposed patients. Although the molecular mechanisms responsible for the etiology of most diseases are unknown, there is evidence of both genetic and environmental factors that could influence this development. Recent findings involve epigenetic mechanisms in the origin of various diseases. This review aims to describe in detail the mechanisms of epigenetic regulation and the known findings involving the dysfunction of these mechanisms as a possible cause of various diseases.
Key words: Epigenetics; Gene expression; Environment
El genoma humano contiene aproximadamente 30 000 genes; sin embargo, cada tipo de célula sólo expresa una pequeña cantidad de genes necesarios para su funcionamiento adecuado. Por otra parte, especies distantes fenotípica y hasta filogenéticamente mantienen genomas con alto grado de homología. Luego de las expectativas puestas en el "proyecto genoma humano", se ha observado que sólo un reducido grupo de enfermedades se puede predecir por la presencia de tal o cual característica genética (mutación). Vale la pena aclarar que para varias enfermedades se han encontrado factores de riesgo que denotan mayor o menor grado de probabilidad en el desarrollo de las mismas sin ser determinantes de su aparición. Además de su genoma, cada célula tiene un epigenoma, el cual es definido por el perfil de expresión génica en respuesta a estímulos ambientales y mecanismos regulatorios a nivel molecular. El término epigenética fue introducido por Conrad Waddington a principios de 19401-3, definiéndola como "la rama de la biología que estudia las interacciones causales entre los genes y sus produtos que dan lugar al fenotipo"2.
En la actualidad, se puede definir a la epigenética como aquellos cambios estables y heredables en la expresión génica, que no son atribuidos a alteraciones en las secuencias de ADN3. Estos cambios pueden afectar tanto la expresión de los genes como las propiedades de sus productos finales.
Para comprender mejor este concepto, podemos decir que la regulación de la expresión génica tiene dos componentes principales2: el primero está determinado por el control inmediato que ejercen activadores y represores de la trascripción sobre los genes. El segundo componente encargado de regular la expresión génica, está determinado por los controles epigenéticos que se ejercen sobre la expresión de la misma2. Los mecanismos epigenéticos que actúan regulando la trascripción incluyen principalmente: a) la metilación del ADN; b) cambios en la configuración de la cromatina; c) la impronta génica; d) el silenciamiento de los genes por unión de ARN3-6. En conjunto, estos cuatro mecanismos epigenéticos junto con el control ejercido por activadores y represores, tienen la capacidad de regular qué genes se expresarán y en qué momento de la vida de la célula.
Dado que la mayor parte de las enfermedades neurológicas son esporádicas, es muy difícil identificar los genes responsables de su desarrollo. Por otro lado, se han podido asociar diferentes genes al desarrollo de enfermedades neurológicas; el hecho de estar sólo asociados indica que no son la causa de las enfermedades sino simplemente marcadores de riesgo. De forma contraria, va en aumento el número de factores ambientales que han sido identificados y relacionados con el desarrollo de diferentes enfermedades neurológicas. La tendencia actual es considerar que los cambios ambientales influyen en el desarrollo de enfermedades a través de cambios en mecanismos epigenéticos.
Por lo previamente considerado y dada la importancia del concepto epigenético en diversas enfermedades, el presente trabajo tuvo como objetivo revisar el rol de la regulación epigenética en el desarrollo de las enfermedades y su posible aplicación terapéutica.
Los mecanismos epigenéticos
Determinamos que la epigenética consiste en un componente fundamental de regulación de la expresión génica. Los mecanismos epigenéticos utilizados con este fin, como previamente mencionamos son: a) la metilación del ADN; b) cambios en la configuración de la cromatina; c) la impronta génica; d) el silenciamiento por unión de ARN3-7.
a) Metilación del ADN
De las 4 bases del ADN, solo la citosina es fisiológicamente modificada a un análogo llamado 5´metilcitosina por la adición de un grupo metilo en el carbono 5 del anillo de pirimidina. Las enzimas responsables de la metilación del ADN son las DNMTs (ADN metiltransferasas) que son divididas en grupos de acuerdo al ADN que utilizan como sustrato: A) metiltransferasas de novo (DNMT3a y DNMT3b) son responsables principalmente de agregar grupos metilos a citosinas en regiones CpG que no habían sido metiladas con anterioridad, B) la metiltransferasa de mantenimiento (DNMT1) copia patrones de metilación preexistentes en cadenas de ADN nuevas durante la replicación. La metilación de las citosinas ayuda a mantener la integridad del genoma al generar el silenciamiento y la imposibilidad de expresarse del gen sobre el que se adicionó el grupo metilo. El mencionado evento en las islas CpG, reprime la trascripción génica, tanto en condiciones fisiológicas como de enfermedad2, 14-18.
Las islas CpG son regiones cromosómicas donde están agrupados sitios CpG no metilados en el extremo 5´ de un gen formando parte de su promotor. Existen excepciones a esta definición, ya que algunos genes poseen esta región metilada, por ejemplo en uno de los cromosomas X de las mujeres, permitiendo su inactivación generando el efecto de compensación de dosis. Los dinucleótidos CpG que no están incrustados en las islas CpG se encuentran generalmente metilados y están esencialmente localizados en secuencias repetitivas o centroméricas, pudiendo además estar distribuidos en regiones génicas e intergénicas.
Por otro lado, las enzimas DNMTs jugarían un rol noenzimático en el silenciamiento transcripcional, influyendo en la remodelación de la cromatina al interaccionar bioquímicamente con las histonas-metiltransferasas y con las histonas-desacetilasas.
Es importante señalar que la represión transcripcional no es el único evento mediado por la metilación del ADN. Se ha informado también que la metilación del ADN está asociada al mantenimiento de la integridad del genoma mediante la supresión de la recombinación mitótica y/o contribuyendo a la cuidadosa segregación de los cromosomas durante la mitosis. Inversamente, se ha descrito que cuando el ADN se encuentra hipometilado en zonas de alto riesgo de mutación, éste sufre una menor perdida de genes, un menor silenciamiento génico y consecuentemente un mayor riesgo de mutación15, 16.
A nivel molecular, existirían 2 mecanismos principales por los cuales la metilación del ADN inhibiría la expresión génica: 1) la modificación de la citosina inhibiría la unión de factores activadores de la trascripción sobre el ADN, impidiendo así la expresión génica; 2) una serie de proteínas que tienen la capacidad de reconocer y unirse a la 5´metilcitosina (MBDs) por medio de diferentes dominios proteicos (MeCP2, MBD1, MBD2, MBD3 y MBD4) ejerciendo su capacidad represiva sobre la expresión del ADN metilado por mecanismos aún no del todo aclarados19. De esta manera, la metilación del ADN tendría un rol sumamente importante en la regulación de la expresión génica (Fig. 1).
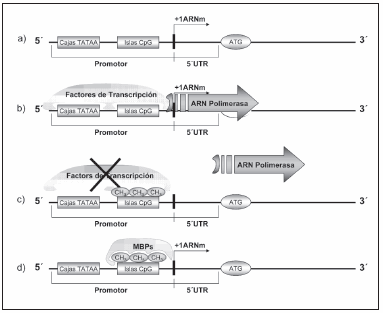
Fig. 1.- Inhibición de la transcripción génica por medio de metilación. a) Esquema de un promotor clásico. b) La unión de los factores de trascripción a los elementos promotores permite que se ubique correctamente la ARN polimerasa y comience con la trascripción. c) la adición de grupos metilos a las citosinas impide la unión de los factores de transcripción inhibiendo la transcripción. d) la unión de proteínas que reconocen metilcitosina (MBDs) inhiben la trascripción.
b) Cambios en la configuración de la cromatina
El nucleosoma es la unidad fundamental de la comatina. Está compuesto por un octámero de histonas (H2A, H2B, H3 y H4) rodeado por dos vueltas de ADN (146pb). Cada nucleosoma está unido con el siguiente por un pequeño segmento de ADN. El nucleosoma puede encontrarse condensado impidiendo la expresión génica o relajado permitiendo la transcripción. Esa variabilidad en la arquitectura del nucleosoma y su estado de relajación o condensación, es regulada por enzimas nucleares que molecularmente tienen la capacidad de modificar el estado de acetilación de lisinas en la cola aminoterminal de las histonas. Estas enzimas son conocidas como histonas desacetilasas (HDACs) producen la desacetilación de las histonas y consecuentemente la condensación del nucleosoma impidiendo la transcripción génica. Por otro lado, el grupo de enzimas histonas acetiltransferasas (HATs) producen la hiperacetilación de las histonas y consecuentemente la relajación del nucleosoma, favoreciendo así la transcripción génica3, 7, 15, 17-19. Ambos grupos de enzimas (HDACS y HATs) no sólo están implicados en el estado de acetilación de las histonas y regulación de la transcripción, sino que también tienen la capacidad de producir interacciones del tipo proteína-proteína, jugando un rol crítico en muchos procesos celulares incluyendo la progresión del ciclo celular, diferenciación y apoptosis17, 20.
Las histonas sufren cambios una vez traducidas. Esas modificaciones post-traduccionales de las histonas nucleosomales -tanto a nivel local como genómico, están siempre implicadas en la regulación transcripcional. Esta es la hipótesis del llamado código de las histonas, a través del cual se explica como diferentes modificaciones amino terminales de las histonas pueden generar interacciones sinérgicas o antagónicas de las proteínas asociadas a la cromatina, regulando de esta manera el acceso al ADN subyacente y a la transcripción15, 16, 19. Funcionalmente, podemos resumir sobre este mecanismo regulatorio, que el genoma eucariota está dividido en eucromatina y heterocromatina, las que son citológica, genómica y funcionalmente distintas. La eucromatina corresponde en general a regiones genómicas que poseen genes transcripcionalmente activos (o potencialmente activos) y que son descondensados durante la interfase. La secuencia regulatoria en estas regiones es accesible a nucleasas y está hiperacetilada sobre los residuos aminoterminales de lisinas. En general, los dominios eucromáticos replican temprano en fase S2, 15, 16.
Mientras que se define a la heterocromatina por su condensación genómica, como una zona transcripcionalmente inactiva, la heterocromatina está silenciada a través de mecanismos epigenéticos (hipoacetilación y metilación de histonas) impidiendo de esta manera la expresión génica en un momento determinado11, 15, 21, 45. En este punto vale la pena aclarar que la metilación de residuos de histonas no siempre está relacionada con la inactivación de la transcripción. Las enzimas encargadas de metilar algunos de los residuos de las histonas son las histonas metiltransferasas (HMTs), que principalmente tienen una alta afinidad por los residuos arginina o lisina; a esta última se la puede encontrar mono, bi o trimetilada. La metilación de H3K4 es una marca constitutiva de eucromatina. Por otro lado, en un reciente trabajo se ha relacionado la metilación de H3K4 en promotores con bajos contenidos de CpG con un alto nivel de expresión génica en células T humanas. La metilación de arginina tanto en la H3 como en la H4 dirigen al proceso de activación transcripcional; de forma contraria, la metilación de H3K9 se asocia mayormente con la formación de heterocromatina15, 46, 47. Se ha observado que la desmetilación de H3K4 por las desmetilasas específicas de lisinas (LSD1 y LSD2) está relacionada con la metilación del ADN dependiente de Dnmt3a-Dnmt3L47.
c) Impronta génica
En humanos, existen varios genes bialélicos sobre los cuales se produce la expresión de un alelo parental, pudiendo ser el paterno o el materno pero nunca ambos. La expresión normalmente se produce en algunas células (exclusión alélica), aunque la secuencia de ambos alelos parentales sea normal33. En algunos casos, la elección de cuál de las dos copias heredadas será expresada no es azarosa, siendo el alelo que siempre es heredado el paterno, mientras que en otros casos el alelo que es expresado es el materno. Este proceso es conocido como impronta genética (imprinting), definida como aquella modificación reversible de la actividad génica dependiendo del sexo parental del cual es transmitido el alelo determinado. El mantenimiento de la impronta genética se da por medio de la metilación del ADN alelo específico, permitiendo que la impronta genómica sea tejido específica o tipo celular específica22, 24.
d) Silenciamiento por unión de ARN
El concepto tradicional es que los genes codifican proteínas a través de una molécula de ARNm intermediario. Evidencias recientes han demostrado que existen moléculas de ARN que no codifican proteínas (ncARNs, del inglés nonprotein-coding RNAs). Esas moléculas tendrían su origen en segmentos de ADN intrónico de genes codificantes de proteínas así como en intrones y exones de genes codificantes de productos no proteicos2, 23. Dichos nc ARNs tendrían la capacidad de regular la expresión génica de muchas maneras, entre ellas afectando la trascripción, la cantidad de ARNm, la traducción y el remodelamiento de la cromatina. Surge de esta manera el concepto de ARNs transcriptos en forma antisentido, (ncARNs tales como ARNi) o ARNs de interferencia (ARNi, ARN de doble cadena capaces de inducir procesos epigenéticos de silenciamiento génico a través de la degradación de los ARNm homólogos, resultando en la pérdida de producción de proteína)23, implicados en el control de la arquitectura cromosómica, en los niveles de ARNm, en la regulación, trascripción y en el corte-empalme alternativo (alternative splicing) de las secuencias de ARNm. De esta manera, los ncARNs serían un mecanismo epigenético involucrado en el control de la expresión génica23.
A través de los cuatro mecanismos previamente explicados, se produciría un control epigenético de la expresión génica celular, regulando de esta manera su función y diferenciación.
Epigenética y enfermedades neurológicas
Las investigaciones realizadas en cáncer, permitieron identificar la contribución de la epigenética en el desarrollo de varias enfermedades humanas. Las enfermedades neurológicas, particularmente aquellas de complejidad genética y clara influencia ambiental, están siendo cada vez más consideradas desde un punto de vista epigenético4 (Tabla 1). Incluso a nivel experimental, parecería que la modulación epigenética contribuiría al control de algunos signos.
TABLA 1.- Enfermedades neurológicas asociadas con factores epigenéticos

Síndrome Rubinstein-Taybi (RTS)
Es un síndrome autosómico dominante, tiene una incidencia de 1:125 000 y se caracteriza por un componente importante de déficit cognitivo. El gen que codifica para la proteína ligando de CREB (CBP) es un co-activador transcripcional que posee actividad acetiltransferasa. Se han propuesto diferentes mutaciones en el dominio HAT de dicho gen, como responsables del desarrollo de RTS, permitiendo inferir que dichas mutaciones alteran productos génicos que estarían implicados en procesos epigenéticos que desencadenarían el desarrollo de la enfermedad.
Se ha concluido en que el desarrollo de RTS está asociado a desregulaciones en la configuración de la cromatina24-26. Los ratones deficientes en CBP, presentan alteraciones de la conducta. Aparentemente, la desregulación del gen CBP inhibiría la diferenciación de los precursores corticales de todos los tipos celulares. Esto coincide con alteraciones de CBP y la acetilación de las histonas a nivel de los promotores de genes proneurales.
En los modelos animales heterocigotas, la administración de inhibidores de la desacetilación produce la restauración de las habilidades de aprendizaje y plasticidad sináptica que estos animales presentan27.
Síndrome de Rett
El síndrome de Rett se caracteriza por graves trastornos en el desarrollo neurológico y la sintomatología predominante está asociada a trastornos cognitivos. Su incidencia es de 1 por cada 15 000 niñas nacidas. Desde el punto de vista genético el síndrome de Rett ha sido asociado con mutaciones en los genes MeCP2 y CdKL5 del cromosoma X48, 49. El gen MeCP2 produce proteínas de unión a metil-CpG, cumpliendo un rol importante en la regulación de la expresión de otros genes y en la plasticidad sináptica28. Los antidepresivos y las drogas de abuso inducen la expresión del factor epigenético MeCP2, influyendo en la remodelación de la cromatina. En trabajos experimentales se ha demostrado que el aumento de los niveles de MeCP2 da lugar a la represión de CdKL5 en las estructuras cerebrales de ratas que habían sido tratadas con cocaína; el mismo resultado se observó en ratas expuestas a serotonina o con sobrexpresión de MeCP2. Por otra parte, se confirmó la regulación de la expresión de CdKL5 a través de la expresión de MeCP2 al observar un aumento en la expresión de CdKL5 cuando es inhibida la expresión de MeCP2 por ARNi, o cuando se tratan las células con inhibidores de ADN-metiltransferasas. En el mismo trabajo se demostró que la metilación de CdKL5 y su metilación dependiente de unión de MeCP2 aumentaron en el striatum de ratas tratadas con cocaína29.
Los ARNmi (microARNs) son regulados por procesos como metilación del ADN. Esta marca epigenética es reconocida por reguladores de la trascripción tales como MeCP2. En un trabajo reciente se estudió la deficiencia en la regulación de ARNmi causada por la ausencia de MeCP2 en un modelo murino de síndrome de Rett, obteniéndose como resultado cambios en los niveles de expresión de ARNmi, que influirían en los niveles de expresión de Irak130. Por otro lado, se ha estudiado la posibilidad de utilizar vectores con el gen MeCP2 como posibles terapias contra el síndrome de Rett. Si bien los resultados experimentales fueron positivos, falta un tiempo aún para que la terapia génica se convierta en un tratamiento aprobado para esta enfermedad31.
Corea de Huntington
Es una enfermedad neurodegenerativa, autosómica dominante, con una penetrancia del 100% y una incidencia de 1-5:100 000. Se caracteriza por desórdenes del movimiento y trastornos cognitivos progresivos. Desde el punto de vista molecular se caracteriza por mutaciones en el gen de la proteína huntingtina (cromosoma 4). Al aumentar el número de repeticiones CAG (35-250) se generan interacciones anormales entre la proteína huntingtina y otras proteínas, conduciendo de manera no aclarada hasta el momento, al desarrollo de la enfermedad. Un dato interesante de la enfermedad es que la edad de comienzo de la misma está relacionada con el número de repeticiones. En varios trabajos se ha observado que las mutaciones en el gen de la huntingtina influyen a nivel nuclear en la expresión de otros genes en las neuronas al inhibir la función de acetilación de histonas realizada por CBP. Experimentalmente se puede contrarrestar este efecto usando inhibidores de la desacetilación. Es importante mencionar que otros trabajos no pudieron replicar estos resultados32-36.
Por otra parte, un gran número de trabajos sugieren el uso de ARNi como posible terapia para la corea de Huntington y se estudia la posibilidad de que el tratamiento sea alelo específico debido a que la mayoría de los pacientes son heterocigotas para la mutación, de manera que se podría disminuir en forma selectiva la expresión de la variable proteica aberrante.
Ataxia de Friedreich
Es una enfermedad autosómica recesiva, que afecta a 1-3 de 100 000 niños recién nacidos. Se presenta como un desorden neurodegenerativo grave. Es causada por mutaciones de repeticiones numéricas de GAA en el gen FXN (proteína mitocondrial frataxina) reduciendo su expresión. Se han propuesto dos modelos para tratar de explicar este cambio en la expresión génica: el primero propone que al producirse un aumento en el número de repeticiones, el ADN adopta una conformación tridimensional diferente impidiendo la progresión de la ARN polimerasa reduciendo la transcripción génica; el segundo hace referencia a que el aumento en el número de repeticiones del triplete induce a la heterocromatización, al producirse una hipoacetilación de las histonas H3-H4, y a un aumento en la metilación de H3K937-39.
Se ha demostrado en diferentes trabajos que el inhibidor de HDCA (HDACIs) aumenta la expresión de FXN en modelos murinos. Sin embargo, no se obtiene el mismo resultado al administrar otros HADCIs como SAHA (del inglés, Suberoylanilide hydroxamic acid) o TSA (del inglés, sodium butyrate trichostatin A)40, 41.
Enfermedad de Alzheimer
Es la causa más común de demencia en edad avanzada. Es una enfermedad neurodegenerativa, que se manifiesta como deterioro cognitivo y trastornos conductuales. Se caracteriza en su forma típica por una pérdida progresiva de la memoria y de otras capacidades mentales. El mayor riesgo asociado es el avance de la edad. Si bien se han detectado variables genéticas de asociación, éstas no son significativas ni determinantes, por lo que se está estudiando el componente epigenético en el inicio y desarrollo de la enfermedad42. En modelos experimentales de enfermedad de Alzheimer, la administración de inhibidores de HDAC, revertiría algunos de los déficit cognitivos43. Obviamente, gran parte de las investigaciones se centran en la modulación de la producción de APP (del inglés, Amyloid Precursor Protein)50. En modelos animales de la enfermedad se han conseguido resultados muy alentadores con el tratamiento con ARNi alelo específicos en el hipocampo, logrando una importante mejoría cognitiva44.
Concluimos en que gran parte de las enfermedades neurológicas carecen de una explicación de sus mecanismos moleculares específicos de producción. Parecería que la "llave" de entrada para la comprensión de estos mecanismos está en el conocimiento de la relación entre la información genética del individuo y las características ambientales capaces de modular la expresión de genes patogénicos o el silenciamiento de genes protectores. En la actualidad existen evidencias que indican que los factores ambientales producirían cambios fisiológicos a través de mecanismos epigenéticos que desencadenarían diferentes enfermedades en individuos genéticamente predispuestos. La comprensión de los mecanismos epigenéticos cobra importancia ya que permitiría el desarrollo de fármacos mecanismo-específicos. Sin embargo, el nivel de conocimiento de los mecanismos moleculares implicados es aún relativamente bajo; por lo tanto, es de suma importancia ahondar en la investigación de estos procesos.
Conflictos de interés: Los autores no presentan conflictos de interés relacionados con este trabajo de revisión.
1. Waddington CH. Selection of the genetic basis for an acquired character. Nature 1952; 169: 625-6. [ Links ]
2. Dupont C, Armant DR, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med 2009; 27: 351-7. [ Links ]
3. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429: 457-63. [ Links ]
4. Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol 2009; 8: 1056-72. [ Links ]
5. Dolinoy DC, Jirtle RL. Environmental epigenomics in human health and disease. Environ Mol Mutagen 2008; 49: 4-8. [ Links ]
6. Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 2005;102: 10604-9. [ Links ]
7. Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood 2009;114: 3727-35. [ Links ]
8. Shiraishi M, Oates AJ, Sekiya T. An overview of the analysis of DNA methylation in mammalian genomes. Biol Chem 2002; 383: 893-906. [ Links ]
9. Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 2006; 31: 89-97. [ Links ]
10. Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene 2008; 27: 5869-85. [ Links ]
11. Lewis J, Bird A. DNA methylation and chromatin structure. FEBS Lett 1991; 285: 155-9. [ Links ]
12. Floess S, Freyer J, Siewert C, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 2007; 5: e38. [ Links ]
13. Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor- induced FoxP3 gene expression: a role for DNA methylation. J Exp Med 2007; 204: 1543-51. [ Links ]
14. Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer's disease. PLoS One 2009;4: e6617. [ Links ]
15. Quina AS, Buschbeck M, Di Croce L. Chromatin structure and epigenetics. Biochem Pharmacol 2006; 72: 1563-9. [ Links ]
16. Waggoner D. Mechanisms of disease: epigenesis. Semin Pediatr Neurol 2007; 14: 7-14. [ Links ]
17. Gray SG, Dangond F. Rationale for the use of histone deacetylase inhibitors as a dual therapeutic modality in multiple sclerosis. Epigenetics 2006;1: 67-75. [ Links ]
18. Heyse KS, Weber SE, Lipps HJ. Histone modifications are specifically relocated during gene activation and nuclear differentiation. BMC Genomics 2009; 10: 554. [ Links ]
19. Lee CG, Sahoo A, Im SH. Epigenetic regulation of cytokine gene expression in T lymphocytes. Yonsei Med J 2009; 50: 322-30. [ Links ]
20. Hewagama A, Richardson B. The genetics and epigenetics of autoimmune diseases. J Autoimmun 2009; 33: 3-11. [ Links ]
21. Peng JC, Karpen GH. Epigenetic regulation of heterochromatic DNA stability. Curr Opin Genet Dev 2008; 18: 204-11. [ Links ]
22. Jirtle RL, Sander M, Barrett JC. Genomic imprinting and environmental disease susceptibility. Environ Health Perspect 2000; 108: 271-8. [ Links ]
23. Pushparaj PN, Melendez AJ. Short interfering RNA (siRNA) as a novel therapeutic. Clin Exp Pharmacol Physiol 2006; 33: 504-10. [ Links ]
24. Bentivegna A, Milani D, Gervasini C, et al. Rubinstein-Taybi Syndrome: spectrum of CREBBP mutations in Italian patients. BMC Medical Genetics 2006, 7: 77. [ Links ]
25. Roelfsema JH, White SJ, Ariyürek Y, et al. Genetic Heterogeneity in Rubinstein-Taybi Syndrome: Mutations in Both the CBP and EP300 Genes Cause Disease. Am J Hum Genet 2005; 76: 572-580. [ Links ]
26. Murata T, Kurokawa R, Krones A, et al. Defect of histone acetyltranferase activity of the nuclear transcripcional coactivator CBP in Rubintein-Taybi syndrome. Humman Molecular Genetics 2001; 10: 1071-1076. [ Links ]
27. Wang J, Weaver IC, Gauthier-Fisher A, et al. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev Cell 2010; 18: 114. [ Links ]
28. Gibson JH, Slobedman B, Karikrishnan NH, et al. Downstream targets of methyl CpG binding protein 2 and their abnormal expression in the frontal cortex of the human Rett syndrome brain. BMC Neurosci 2010; 11: 53. [ Links ]
29. Carouge D, Host L, Aunis D, Zwiller J, Anglard P. CDKL5 is a brain MeCP2 target gene regulated by DNA methylation. Neurobiol Dis 2010; 38: 414-24. [ Links ]
30. Urdinguio RG, Fernandez AF, Lopez-Nieva P, et al. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 2010; 5. [Epub ahead of print]. [ Links ]
31. Rastegar M, Hotta A, Pasceri P, et al. MECP2 isoformspecific vectors with regulated expression for Rett syndrome gene therapy. PLoS One 2009; 4: e6810. [ Links ]
32. Pérez P C, Miranda C M, Segura-Aguilar J. Availability of genetic diagnosis of Huntington disease in Chile. Rev Med Chil 2009; 137: 1128-9. [ Links ]
33. Swami M, Hendricks AE., Gillis T, et al. Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Human Molecular Genetics 2009; 18: 3039-47. [ Links ]
34. Klevytska AM, Tebbenkamp AT, Savonenko AV, Borchelt DR. Partial depletion of CREB-binding protein reduces life expectancy in a mouse model of Huntington disease. J Neuropathol Exp Neurol 2010; 69: 396-404. [ Links ]
35. Jiang H, Poirier MA, Liang Y, et al. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol Dis 2006; 23: 543-51. [ Links ]
36. Lee JM, Zhang J, Su AI, et al. A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst Biol 2010; 4: 29. [ Links ]
37. Punga T, Buhler M. Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol Med 2010; 2: 120-9. [ Links ]
38. Li K, Singh A, Crooks DR, et al. Expression of Human Frataxin Is Regulated by Transcription Factors SRF and TFAP2. PLoS One 2010; 5: 1-8. [ Links ]
39. Ditch S, Sammarco MC, Banerjee A, Grabczyk E. Progressive GAA.TTC repeat expansion in human cell lines. PLoS Genet 2009; 5: 1-12. [ Links ]
40. Rai M, Soragni E, Chou CJ, et al. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich's ataxia patients and in a mouse model. PLoS One 2010; 5: 1-8. [ Links ]
41. Xu C, Soragni E, Chou CJ, et al. Chemical probes identify a role for histone deacetylase 3 in Friedreich's ataxia gene silencing. Chem Biol 2009;16: 980-9. [ Links ]
42. Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer's disease. PLoS One 2008; 3: e2698. [ Links ]
43. Kilgore M, Miller CA, Fass DM, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. J Psychiatry Neurosci 2010; 35: 870-80. [ Links ]
44. Rodríguez-Lebrón E, Gouvion CM, Moore SA, Davidson BL, Paulson HL. Allele-specific RNAi mitigates phenotypic progression in a transgenic model of Alzheimer's disease. Mol Ther 2009; 17: 1563-73. [ Links ]
45. Allfrey VG, Faulkner R and Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Biochemistry 1964; 51: 786-94. [ Links ]
46. Karlic R, Chung HR, Lasserre J, Vlahovicekb K, and Vingron M. Histone modification levels are predictive for gene expresión. Proc Natl Acad Sci USA 2010; 107: 2926-31. [ Links ]
47. Cheng X, Blumenthal RM. Coordinated chromatin control: structural and functional linkage of DNA and histone methylation. Biochemistry 2010; 49: 2999-3008. [ Links ]
48. Ruggieri V, Arberas C. Trastornos generalizados del desarrollo. Medicina (Buenos Aires) 2007; 67: 569-85. [ Links ]
49. Campos-Castello J, Fernández-Mayoralas DM, Muñoz- Jareño N, San Antonio-Arce V. Síndrome de Rett: 50 años de historia de un trastorno aún no bien conocido. Medicina (Buenos Aires) 2007; 67: 531-42. [ Links ]
50. Leal MC, Fernández Gamba A, Morelli L, Castaño EM. Proteólisis cerebral del péptido amieloide ß: Relevancia de la enzima degradadora de insulina en la enfermedad de Alzheimer. Medicina (Buenos Aires) 2009; 69: 466-72. [ Links ]
Recibido: 16-11-2010
Aceptado: 7-6-2011

