










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680
Medicina (B. Aires) vol.72 no.4 Ciudad Autónoma de Buenos Aires Aug. 2012
COMUNICACIÓN BREVE
El receptor de estrógenos alfa como mediador del efecto proliferativo de progestágenos en cáncer de mama*
Sebastián Giulianelli1, José P. Vaqué2, Victoria Wargon1, Rocío Soldati1, Silvia I. Vanzulli3, Rubén Martins4, Eduardo Zeitlin4, Luisa Helguero5, Caroline Lamb1, Alfredo A. Molinolo2, J. Silvio Gutkind2, Claudia Lanari1
1Instituto de Biología y Medicina Experimental, IByME-CONICET, Buenos Aires;
2Oral and Pharyngeal Cancer Branch, NIDCR, NIH, Bethesda, USA;
3Academia Nacional de Medicina;
4Policlínica Bancaria, Buenos Aires;
5Universidade de Aveiro, Aveiro, Portugal
Dirección postal: Dra. Claudia Lanari, Instituto de Biología y Medicina Experimental, Vuelta de Obligado 2490, 1428 Buenos Aires, Argentina.
Fax: (54-11) 4786-2564 e-mail: lanari.claudia@gmail.com
*Este trabajo fue distinguido con el Premio Cherny en la reunión anual de la SAIC (Sociedad Argentina de Investigación Clínica) en Mar del Plata, noviembre 2011.
Resumen
En carcinomas mamarios murinos (C4-HD) y en células de cáncer de mama humano (T47D) observamos que el progestágeno sintético, acetato de medroxiprogesterona (MPA), induce la activación del receptor de estrógenos alfa (REa) y su asociación nuclear con el receptor de progesterona (RP). En este trabajo postulamos que dicha interacción a nivel genómico sería fundamental para desarrollar respuestas proliferativas mediadas por progestágenos. Demostramos que el antiestrógeno fulvestrant (FUL, ICI182.780) indujo la regresión completa de tumores C4-HD creciendo con MPA. El progestágeno indujo la expresión temprana de CCND1 y MYC en células T47D y este efecto fue revertido al bloquear el REa. En células tratadas con MPA utilizamos ensayos de inmunoprecipitación de la cromatina (ChIP) y corroboramos la colocalización nuclear de RP/REa en los mismos sitios de los promotores de CCND1 y MYC. El ICI no afectó la unión de RP a ambas secuencias regulatorias, pero sí inhibió la unión del REa. Confirmamos la interacción nuclear entre REa y RP en muestras de cáncer de mama humano. Los resultados demuestran que la presencia del REa, interactuando con el RP, en promotores de CCND1 y MYC es fundamental para la transcripción génica y la proliferación celular inducida por el progestágeno.
Palabras clave: Cáncer de mama; REa; RP; MPA; CCND1/MYC
Abstract
The role of estrogen receptor alpha in breast cancer cell proliferation mediated by progestins. In C4-HD murine mammary carcinomas and in human breast cancer T47D cells, we showed that medroxyprogesterone acetate (MPA) induces a nuclear physical association between estrogen receptor alpha (ERa) and progesterone receptors (PR). The blockade of ERa inhibits cell proliferation mediated by progestins. We hypothesized that this nuclear association between ERa/PR is necessary to trigger progestin-induced cell proliferation and tumor growth. We demonstrated that fulvestrant (FUL, ICI182.780) induced complete regression of C4-HD tumors growing with progestins. MPA treatment induced an early increase in both CCND1 and MYC expression in T47D cells. The blockade of ERa prevented the MPA-dependent transcription of both genes. Specific binding of PR/ERa was observed at the same MPA-sensitive regions at the CCND1 and MYC gene promoters after chromatin immunoprecipitation (ChIP) analysis. ICI inhibited binding of ERa to both gene regulatory sequences while PR binding was unaffected. The nuclear colocalization bet ween both receptors in T47D cells was confirmed by: confocal microscopy, Duolink assays and co-immunoprecipitation assays. In breast cancer samples we also observed a nuclear interaction between both steroid receptors. Our results indicate that the presence of ERa interacting with activated PR at the CCND1 and MYC promoters is required to trigger progestin-induced gene transcription and cell proliferation in breast cancer cells.
Key words: Breast cancer; ERa; PR; MPA; CCND1/MYC
Los mecanismos por los cuales los estrógenos y los progestágenos, a través de sus respectivos receptores nucleares [receptores de estrógenos alfa (REa) y receptores de progesterona (RP) respectivamente], participan en el desarrollo y crecimiento del cáncer de mama es un tema controvertido y de gran interés. La detección de estos receptores se utiliza como factor pronóstico y orienta la terapéutica hacia una de tipo hormonal, dirigida principalmente a bloquear la fuente de estrógenos o inhibir la acción del REa. Sin embargo, en los últimos años han surgido numerosas evidencias que implican a los RP en el desarrollo y progresión de carcinomas mamarios1.
En nuestro laboratorio se ha desarrollado un modelo experimental de adenocarcinomas mamarios murinos inducidos por la administración prolongada de acetato de medroxiprogesterona (MPA) en ratones hembras vírgenes de la cepa BALB/c. Se establecieron líneas tumorales hormono-dependientes (entre ellas la C4-HD) que expresan REa, REβ, ambas isoformas del RP (RPA y RPB) y que crecen in vivo solo en presencia de MPA o progesterona (Pg)2. Hemos demostrado que el RP es fundamental en el crecimiento de dichos tumores, dado que el tratamiento con antiprogestágenos (mifepristona, onapristona, lonaprisan) u oligonucleótidos antisentido para el RP induce regresión tumoral. Paradójicamente, tanto los estrógenos como el antiestrógeno tamoxifeno inhiben el crecimiento tumoral3. Todas estas observaciones nos llevaron a investigar qué papel cumple el REa, además del RP, en el crecimiento de dichos tumores.
Está ampliamente aceptado que el RP es un gen regulado por estrogenos4 y, considerando que dicho receptor es usualmente utilizado como marcador de un REa activo, sería intuitivo pensar que hay un efecto secuencial en el REa induciendo la expresión del RP. Por otro lado, se postuló una rápida interacción citoplasmática en células de cáncer de mama humano, T47D, entre el REa y el RPB necesaria para activar la vía de señalización c-Src/ p21ras/Erk por progestágenos5, lo que luego resultaría en la fosforilación y activación del RP. Aunque los dominios que participan en dicha interacción fueron mapeados6, el grupo de D. Edwards propuso que un motivo rico en prolinas, en la región amino-terminal del RP, es suficiente para activar la vía de c-Src por progestágenos7, de manera que no existe un consenso respecto a los mecanismos involucrados. Sin embargo, ambos modelos coinciden en que estos efectos no genómicos del RP llevarían a un incremento en la expresión de Ciclina D1 (CCND1) por progestágenos a través de la activación de factores de transcripción (fos, jun, Sp1) sobre el promotor de dicho gen.
En nuestro modelo de carcinomas mamarios murinos C4-HD, y en células humanas T47D, hemos observado que el MPA induce la asociación nuclear entre el REa y ambas isoformas del RP. Dicha interacción fue estudiada por microscopía confocal, usando anticuerpos específicos para ambos receptores y realizando un recuento de células que muestran colocalización (Fig. 1A), usando la técnica de Duolink, mediante la cual se detectan puntos fluorescentes solo en el caso de existir colocalización molecular (Fig. 1B, y Fig. 1 suplementaria, esta última está disponible en color en la versión on-line de esta publicación, www.medicinabuenosaires.com) y por técnicas de coinmunoprecipitación (CoIP, Fig. 1C). Además, ambos receptores se encuentran fosforilados formando parte de dichos complejos (no se muestra), lo cual sugiere que podrían estar activos a nivel transcripcional. El bloqueo del REa por tratamiento con el antiestrógeno puro ICI 182.780 (ICI; fulvestrant - Fig. 1D) o con ARN de interferencia (siRNA - no se muestra), inhibe la proliferación celular mediada por el progestágeno en células C4-HD3 y T47D. Postulamos que dicha interacción a nivel genómico entre ambos receptores hormonales sería fundamental para desarrollar respuestas proliferativas mediadas por MPA. Nuestro objetivo fue estudiar in vivo la acción del fulvestrant (FUL) sobre el crecimiento de tumores C4-HD estimulados con MPA, los mecanismos moleculares involucrados en la interacción de ambos receptores en células humanas y evaluar dicha interacción en 15 muestras de cáncer de mama humano.
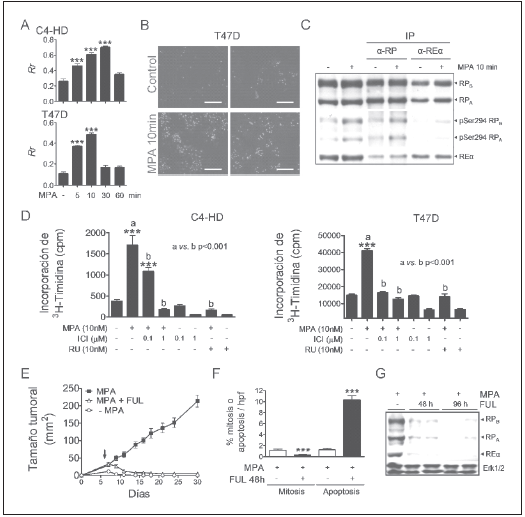
Fig. 1.- El MPA induce la interacción nuclear entre el REa y el RP, mientras que el FUL inhibe el crecimiento tumoral dependiente de MPA. (A) Células epiteliales C4-HD o T47D arrestadas en 1% suero fetal bovino libre de hormonas (SFBch) por 24 h y tratadas con 10 nM de MPA por diferentes tiempos, se procesaron para inmunofluorescencia (IF) y microscopía confocal con anticuerpos específicos anti-RP (Ab7-monoclonal) y anti-REa (MC-20 o SP1-policlonales). La colocalización nuclear entre ambos receptores fue cuantificada estimando el coeficiente de correlación de Pearson (Rr, PSC Colocalization plug-in, ImageJ) en 200 núcleos a 600X. Valores de Rr entre -1 (correlación negativa perfecta) y +1 (correlación positiva perfecta), con 0 indicando nada de correlación. *** p < 0.001. (B) Células T47D incubadas o no por 10 min con MPA fueron procesadas para evaluar la interacción nuclear entre REa (anticuerpo HC-20) y RP (anticuerpo Ab7) utilizando el kit Duolink (Duolink II Orange Starter Kit 92102, Olink Bioscience). Barra: 15 µm. (C) Extractos nucleares de células T47D, tratadas o no con MPA por 10 min se inmunoprecipitaron (IP) con anticuerpos anti-RP (Ab7) o anti-REa (SP1) y se procesaron para SDS-PAGE y Western blot. (D) Ensayos de proliferación por incorporación de 3H-timidina en células C4-HD y T47D. *** p < 0.001. (E) Ratones hembra BALB/c transplantados con tumores C4-HD en presencia de MPA (depot de 20 mg) fueron tratados con FUL (flecha) cuando los tumores alcanzaron un tamaño menor a 50 mm2. Se registró la efectividad del tratamiento durante 23 días y se gr aficó el tamaño tumoral (ancho x largo). A un grupo de ratones se les removió el MPA, demostrando la hormono-dependencia de dicho tumor. (F) Recuento de mitosis y apoptosis luego de 48 h de tratamiento con FUL en tumores C4-HD creciendo con MPA. Se registró el número de mitosis o apoptosis en relación al número total de células en 10 campos de mayor aumento (hpf, 1000X). *** p < 0.001. (G) Extractos totales de tumores C4-HD creciendo con MPA y tratados por 48 o 96 h con FUL fueron procesados para SDS-PAGE y WB con anticuerpos anti-RP (C19) o anti-REa (MC-20). Se utilizó Erk1/2 como control de carga.
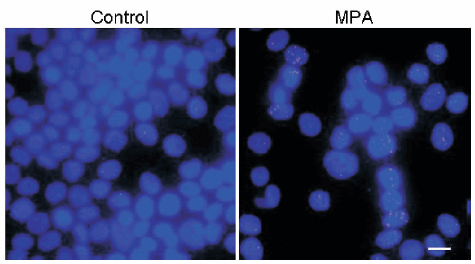
Fig. 1 Suplementaria: Células T47D incubadas o no por 10 min con MPA (10 nM) fueron procesadas para evaluar la interacción nuclear entre REa (anticuerpo HC-20) y RP (anticuerpo Ab7) utilizando el kit Duolink (Duolink II Orange Starter Kit 92102, Olink Bioscience). En azul se ven los núcleos y el puntillado muestra los sitios de colocalización de ambas proteínas. Barra: 15µm
El tratamiento con FUL (5 mg/ semana) durante 23 días indujo la regresión completa de tumores C4-HD creciendo in vivo con MPA (Fig. 1D). A las 48 horas de tratamiento con FUL, observamos una disminución significativa en marcadores de proliferación con respecto a los controles creciendo con MPA: índice mitótico (Fig. 1E), Ki67 (p < 0.001, no se muestra). Asimismo, tanto la expresión del REa como del RP, evaluado por Western blot (WB - Fig. 1F) e inmunohistoquímica (IHQ - no se muestra), disminuyó significativamente (p < 0.001) luego de dicho tratamiento. Además, demostramos que la regresión tumoral por FUL va acompañada por un aumento de apoptosis (Fig. 1F), asociado con una disminución en la expresión BCL/XL y a un aumento en la expresión de BAX y AIF (no se muestra).
Cuando se removió el MPA de animales portadores de tumores C4-HD, observamos a las 48 horas una reducción en la expresión del REa y un aumento significativo del RP (p < 0.001, no se muestra), indicando que mientras que el MPA regula negativamente los niveles del RP, sería responsable de mantener alta la expresión del REa en dichos tumores in vivo. Asimismo, habíamos demostrado que en los tumores C4-HD creciendo con MPA los REa presentan altos niveles de fosforilación (aumento de pSer118 y pSer167 REa, en comparación a tumores donde el MPA fue removido por 48 horas; SAIC 2007), indicando que podrían estar activos a nivel transcripcional.
En células T47D transfectadas con un vector de expresión de luciferasa (Luc) bajo el control de secuencias PRE (progesterone responsive element), demostramos que el ICI (1µM) inhibió la expresión de Luc inducida por 10 nM de MPA (p < 0.001 - no se muestra). Dicha inhibición, no estuvo asociada a una disminución en la expresión del RP por el tratamiento con ICI, dado que observamos alta expresión del receptor luego de 24 horas de incubación con el antiestrógeno (no se muestra). Además, el MPA fue capaz de inducir la expresión de PRE-Luc en células MDA-MB-231 (RP-/REa-) establemente transfectadas con RPB, y dicha inducción fue aún mayor (p < 0.001) en presencia del REa (no se muestra). Estos resultados sugieren fuertemente la participación de complejos REa-RP en elementos regulatorios de genes regulados por MPA.
Asimismo, estudiamos la presencia de dichos complejos en los promotores de CCND1 y MYC, dos genes regulados por MPA8, 9. Observamos una alta expresión de las proteínas CCND1 y MYC en tumores C4-HD creciendo con MPA (no se muestra). Dicha expresión fue inhibida luego de 48 horas de tratamiento con FUL (Fig. 2A). Por otro lado, la expresión de ambos genes (ARN mensajero) fue evaluada por PCR cuantitativa (qPCR) en células T47D, demostrando que el MPA (10 nM) incrementó, a tiempos cortos, los niveles de expresión de CCND1 y MYC (Fig. 2B). El bloqueo del REa por ICI (Fig. 2C) o utilizando un siRNA especifico (no se muestra) que redujo los niveles del receptor evaluado por WB, inhibió la transcripción de ambos genes inducida por el progestágeno. Asimismo, comprobamos la importancia de CCND1 en la proliferación por MPA, utilizando un siRNA específico contra dicho mRNA (no se muestra). Los resultados sugieren que la actividad del REa, presumiblemente a través de su capacidad de interactuar con el RP, puede controlar la expresión de genes asociados a proliferación celular formando complejos nucleares que se unirían a elementos regulatorios y controlarían su expresión en respuesta al MPA.
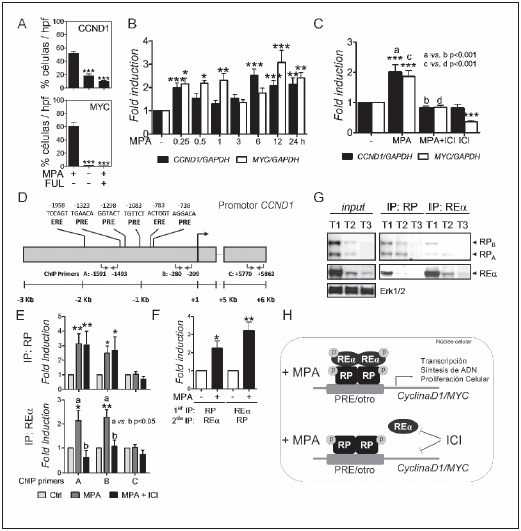
Fig. 2.- El REa y el RP interactúan sobre promotores de CCND1 y MYC, en un mecanismo mediado por MPA. (A) Tumores C4-HD creciendo con MPA y tratados con FUL por 48 h, o en los cuales el depot de MPA fue removido por el mismo período de tiempo, se procesaron para IHQ con anticuerpos específicos anti-CCND1 y MYC. Se cuantificó el número de células con marcación nuclear positiva para cada proteína en función del número de células totales en 10 campos a 1000X (hpf). ***p < 0.001. (B) Los niveles de expresión de ARNm para CCND1 y MYC, relativo a los niveles de expresión de GAPDH, fueron evaluados por qPCR en células T47D luego de tratamiento con MPA (10 nM), *p < 0.05, **p < 0.01, ***p< 0.001. (C) Efecto del ICI (1µM) sobre los niveles de expresión de CCND1 y MYC, evaluado como en (B), inducido por MPA. ***p < 0.001. (D) Esquema del promotor de CCND1 humano, detallando sitios PRE, sitios ERE y primers usados en los experimentos de ChIP (primers A, B y C). Los primers C se utilizaron como control negativo para la unión de los receptores hormonales. (E) Ensayos de ChIP sobre el promotor de CCND1 utilizando anticuerpos anti-RP (H190X), anti-REa (HC20X) o anti-IgG. Células T47D tratadas con 10 nM de MPA o MPA + 1 µM ICI por 10 min, se fijaron en 1% formaldehído, se lisaron y la cromatina sonicada se procesó como se describió previamente10. El ADN purificado luego de la IP se utilizó en ensayos de qPCR (usando la formula 2(-?Ct)) con los primers descriptos en (D). Cada IP fue relativizada a los valores de PCR (Ct) correspondiente a la cromatina que no sufrió IP (inputs), luego a los valores de IgG, y los resultados se muestran como incremento respecto a los valores obtenidos para las células sin tratar con MPA. *p < 0.05, **p < 0.01. (F) Ensayos de ChIP-reChIP en células T47D luego de tratamiento con MPA por 10 min sobre el promotor de CCND1 utilizando anticuerpos anti-RP (H190X), anti-REa (HC20X) o anti-IgG. *p < 0.05, **p < 0.01. (G) Los extractos nucleares de tres tumores humanos (T1, T2 y T3), se inmunoprecipitaron (IP) con anticuerpos anti-RP (Dako) o anti-REa (SP1) y se procesaron para SDS-PAGE y WB. Inputs: extractos nucleares de cada tumor utilizados en las IPs. (H) Modelo propuesto para la interacción genómica entre REa/RP mediada por progestágenos.
Realizamos ensayos de inmunoprecipitación de la cromatina (ChIP) para investigar la presencia del REa y el RP en los promotores de CCND1 y MYC. Para ello, estudiamos la presencia de sitios PRE y ERE (estrogen responsive element) en ambos promotores (solo se muestra para CCND1, Fig. 2D). Está descripto que el promotor de MYC humano posee sitios PRE funcionales8, mientras que para CCND1 no se han informado secuencias PRE o ERE en su promotor. Como se mencionó previamente, la inducción de la expresión de CCND1 por progestágenos sería a través de eventos no genómicos mediados por el RP. Haciendo un análisis minucioso de ambos promotores humanos, encontramos diversos hemi-sitios PRE y ERE en ambas secuencias (solo se muestra para CCND1, Fig. 2D). Dichos elementos podrían mediar la unión de RP y REa activados a ambos promotores, como fue descripto para otros genes8. Observamos que el MPA provocó la unión del RP y el REa a los mismos sitios en el promotor de CCND1 y MYC (solo se muestra para CCND1, Fig. 2E). La unión del RP a ambas secuencias regulatorias no fue afectada por el ICI, el cual inhibió la unión del REa (Fig. 2E). Resultados similares se obtuvieron luego de reducir la expresión del REa con siRNAs específicos (no se muestra). En ensayos de ChIP-reChIP (Fig. 2F), demostramos que ambos receptores ocupan los mismos sitios en los promotores de CCND1 y MYC luego del tratamiento con MPA. Estos resultados sugieren que ambos receptores necesitan interactuar en los promotores de CCND1 y MYC para inducir transcripción génica y proliferación celular en respuesta al MPA, y sustentan la hipótesis de que la presencia del REa en dichos promotores es requerida para inducir los efectos antes descriptos, mediados por el RP.
Por otro lado, realizamos estudios en 15 muestras de cáncer de mama humano, pudiendo confirmar, por microscopía confocal, la colocalización nuclear entre el REa y el RP intensa y moderada en 4 y 2 muestras, respectivamente (no se muestra). Dichos resultados se validaron en ensayos de CoIP a partir de extractos nucleares de tres tumores humanos (Fig. 2G), uno con alta expresión de RP (T1), uno con moderada expresión de RP (T2) y uno RP- (T3), sugiriendo que la interacción nuclear entre ambos receptores podría tener un importante y aun no explorado rol en el cáncer de mama humano.
En este trabajo demostramos que el MPA induce una interacción directa y transitoria entre el REa y ambas isoformas del RP en los promotores de dos genes regulados por progestágenos, CCND1 y MYC. Dicha actividad tendría efectos clave sobre la proliferación de células tumorales y sería dependiente de la actividad del REa, dado que su bloqueo con FUL induce la regresión de tumores C4-HD y la inhibición en la proliferación de células C4-HD y T47D in vitro, mediada por MPA. Nuestros resultados sugieren que la terapia combinada con antiestrógenos y antiprogestágenos podría beneficiar a pacientes seleccionados con cáncer de mama. En modelos experimentales se demostró que el co-tratamiento de antiprogestágenos con moduladores selectivos del REa podría tener efectos aditivos comparado con cada terapia individual10. Más aún, la variante hormono-independiente del tumor C4-HD (C4-HI) responde mejor a la combinación de tamoxifeno con mifepristona (RU-486) que a cada agente por separado11.
Recientemente, demostramos en células T47D que el MPA induce la unión del RP junto con factores de transcripción (TF) como STAT5 y el receptor tirosina quinasa (RTK) FGFR2 a secuencias regulatorias en el promotor de MYC12. Los resultados expuestos en este trabajo indican que el REa activado podría formar parte del mismo complejo multiproteico. Como se explicó previamente, la regulación de CCND1 por progestágenos sería a través de eventos no genómicos mediados por el RP5, 6. Actualmente, la interacción citosólica entre REa y RPB es necesaria para la activación de las vías de señalización c-Src/p21Ras/Erk, PI3K/Akt y JAK/STAT5, 6 por Pg; contrariamente, otro modelo propone que un motivo rico en prolinas en el RP sería suficiente para activar c-Src/Erk o PI3K/Akt7. Nuestro grupo ha demostrado una localización de membrana y citosólica del RP clásico, sin embargo no encontramos una colocalización entre el REa y el RP en dichas estructuras celulares. Ambos modelos suponen que esos mecanismos no genómicos activados por Pg llevarían a la activación de TF en el promotor de CCND1 induciendo la transcripción génica y la proliferación celular. Sin embargo, se ha demostrado recientemente que el RP tendría efectos genómicos sobre el promotor de CCND113, 14 actuando, por ejemplo, como coactivador de STAT315. En este trabajo demostramos por primera vez que el RP y el REa comparten las mismas regiones sensibles al MPA en los promotores de CCND1 y MYC (Fig. 2H). Dichos complejos estarían formados además por coactivadores de receptores hormonales (SRC, steroid receptor coactivator), como por ejemplo AIB1 (SAIC 2010).
Además de haber descripto una interacción genómica entre el REa y el RP, nuestros resultados muestran que las concentraciones antiestrogénicas de ICI fueron capaces de bloquear la formación de dichos complejos, inhibir la transcripción génica y la proliferación celular inducida por MPA, sin afectar en forma temprana la activación del RP. Esto implica un cambio en el paradigma que establece que el REa mediaría la activación de RP por vías no genómicas.
Finalmente, aquí mostramos que ambos receptores hormonales se encuentran asociados en el núcleo en muestras seleccionadas de cáncer de mama humano, sugiriendo que la activación ligando-independiente de dichos receptores podría estar implicada en el crecimiento de dichos tumores. De esta manera, es posible especular que los pacientes con alto grado de colocalización nuclear entre REa-RP podrían tener una mejor respuesta a la terapia combinada de antiestrógenos con antiprogestágenos. El desafío es identificar cuáles son los pacientes que se beneficiarían de estas terapias combinadas.
Conflictos de interés: Los autores declaran no tener conflictos de interés.
1. Lange CA, Sartorius CA, Abdel-Hafiz H, et al. Progesterone receptor action: translating studies in breast cancer models to clinical insights. Adv Exp Med Biol 2008; 630: 94-111. [ Links ]
2. Lanari C, Lamb CA, Fabris VT, et al. The MPA mouse breast cancer model: evidence for a role of progesterone receptors in breast cancer. Endocr Relat Cancer 2009; 16: 333-50. [ Links ]
3. Lamb CA, Helguero LA, Fabris V, Colombo L, Molinolo AA, Lanari C. Differential effects of raloxifene, tamoxifen and fulvestrant on a murine mammary carcinoma. Breast Cancer Res Treat 2003: 79: 25-35. [ Links ]
4. Horwitz KB, Koseki Y, McGuire WL. Estrogen control of progesterone receptor in human breast cancer: role of estradiol and antiestrogen. Endocrinology 1978; 103: 1742-51. [ Links ]
5. Migliaccio A, Piccolo D, Castoria G, et al. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J 1998; 17: 2008-18. [ Links ]
6. Ballaré C, Uhrig M, Bechtold T, Sancho E, et al. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol Cell Biol 2003; 23: 1994-2008. [ Links ]
7. Boonyaratanakornkit V, Scott MP, Ribon V, et al. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol Cell 2001; 8: 269-80. [ Links ]
8. Moore MR, Zhou JL, Blankenship KA, Strobl JS, Edwards DP, Gentry RN. A sequence in the 5' flanking region confers progestin responsiveness on the human c-myc gene. J Steroid Biochem Mol Biol 1997; 62: 243-52. [ Links ]
9. Musgrove EA, Hamilton JA, Lee CS, Sweeney KJ, Watts CK, Sutherland RL. Growth factor, steroid, and steroid antagonist regulation of cyclin gene expression associated with changes in T-47D human breast cancer cell cycle progression. Mol Cell Biol 1993; 13: 3577-87. [ Links ]
10. Nishino Y, Schneider MR, Michna H. Enhancement of the antitumor efficacy of the antiprogestin, onapristone, by combination with the antiestrogen, ICI 164384. J Cancer Res Clin Oncol 1994; 120: 298-302. [ Links ]
11. Wargon V, Helguero LA, Bolado J, et al. Reversal of antiprogestin resistance and progesterone receptor isoform ratio in acquired resistant mammary carcinomas. Breast Cancer Res. Treat 2009; 116: 449-60. [ Links ]
12. Cerliani JP, Guillardoy T, Giulianelli S. Interaction between FGFR-2, STAT5, and Progesterone Receptors in Breast Cancer. Cancer Res 2011; 71: 3720-31. [ Links ]
13. Quiles I, Millán-Ariño L, Subtil-Rodríguez A, et al. Mutational analysis of progesterone receptor functional domains in stable cell lines delineates sets of genes regulated by different mechanisms. Mol Endocrinol 2009; 23: 809-26. [ Links ]
14. Cicatiello L, Addeo R, Sasso A, et al. Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol Cell Biol 2004; 24: 7260-74. [ Links ]
15. Beguelin W, Díaz Flaque MC, Proietti CJ, et al. Progesterone receptor induces ErbB-2 nuclear translocation to promote breast cancer growth via a novel transcriptional effect: ErbB-2 function as a coactivator of Stat3. Mol Cell Biol 2010; 30: 5456-72. [ Links ]
Recibido: 23-4-2012
Aceptado: 17-5-2012

