










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680
Medicina (B. Aires) vol.73 no.1 Ciudad Autónoma de Buenos Aires Feb. 2013
CASUÍSTICA
Osteomalacia inducida por tumor: hemangiopericitoma rinosinusal
Enriqueta M. Serafini, Ana A. Pisarevsky, Javier Plumet Garrido, Rafael J. Zamora, Enrique A. Petrucci
VI Cátedra de Medicina Interna. Servicio de Medicina Interna, Hospital de Clínicas José de San Martín, Universidad de Buenos Aires
Dirección postal: Dra. Ana A. Pisarevsky, VI Cátedra de Medicina Interna, Hospital de Clínicas José de San Martín, Av. Córdoba 2351, 1120 Buenos Aires, Argentina
Fax: (54-11) 4362-2296 e-mail: apisarevsky@gmail.com
Resumen
La osteomalacia inducida por tumor es una rara enfermedad del metabolismo óseo caracterizada por el aumento en la excreción de fosfato a nivel renal seguido de hipofosfatemia. Es causada por agentes fosfatúricos producidos por determinados tumores. La resección total del tumor resulta en la completa reversión de las anormalidades bioquímicas, la desaparición de las manifestaciones clínicas y los hallazgos en los estudios por imágenes. Presentamos el caso de un varón de 61 años con cuadro clínico y laboratorio compatibles con osteomalacia oncogénica inducida por tumor mesenquimático de localización rinosinusal. En nuestro caso el diagnóstico histológico correspondió a una neoplasia de tipo vascular: hemangiopericitoma.
Palabras clave: Osteomalacia inducida por tumor; Hipofosfatemia; Hemangiopericitoma; Osteomalacia oncogénica
Abstract
Tumor-induced osteomalacia: rhinosinusal hemangiopericytoma. Tumor-induced osteomalacia is a rare disease of bone metabolism. The characteristic of this disease is an increase in phosphate excretion followed by hypophosphatemia, due to phosphaturic agents produced by different types of tumors. Tumor resection results in complete resolution of clinical, biochemical and radiological abnormalities. We present the case of a 61 year old man with signs, symptoms and laboratory findings consistent with oncogenic osteomalacia due to a rhino-sinusal mesenchymal tumor. The histological diagnosis showed a vascular neoplasm: hemangiopericytoma.
Key words: Tumor-induced osteomalacia; Hypophosphatemia; Hemangiopericytoma; Oncogenic osteomalacia
La osteomalacia inducida por tumor (OIT), también llamada osteomalacia oncogénica, es un desorden óseo metabólico poco frecuente2 descripto dentro de los síndromes paraneoplásicos, que se caracteriza por una historia de dolores óseos difusos y fracturas que llevan al paciente a la postración1-8. En el laboratorio se destaca como dato cardinal la hipofosfatemia que se debe a una disminución de la reabsorción de fosfato a nivel tubular renal acompañado de una disminución de los niveles plasmáticos de 1.25 dihidroxi-vitamina D3, lo que conduce al cuadro de osteomalacia2, 5.
La OIT se desarrolla a partir de tumores en su mayoría de origen mesenquimático1-8, habitualmente de comportamiento benigno, de lento crecimiento y de difícil detección debido a su tamaño reducido2, 6, 7. Estos tumores fueron descriptos por primera vez en 1947 por McCance, y su asociación con osteomalacia fue informada en 1959 por Prader y col.2. La resección exitosa de los mismos es curativa5, 6. La entidad histológica predominante dentro de los OIT es el hemangiopericitoma2, 4, 7, 8.
Caso clínico
Un hombre de 61 años fue derivado a nuestra institución para evaluación de dolores óseos que le imposibilitaban la marcha. El paciente se encontraba en buen estado de salud hasta 3 años antes de la consulta, cuando comenzó con dolor progresivo de tobillos, rodillas y costillas. Tenía dificultad para caminar, pararse y sentarse. En este período presentó fractura de cadera derecha, fracturas costales y de vértebra dorsal a nivel de D8. Un centellograma corporal total demostró múltiples áreas de captación patológica en todo el esqueleto compatible con secundarismo. Fue evaluado en varios hospitales y con el diagnóstico de metástasis óseas generalizadas con tumor primario desconocido inició tratamiento paliativo. No refería historia familiar de osteopatías, enfermedades metabólicas o reumatológicas. Negaba pérdida de peso, diarrea, abuso del alcohol, uso de antiácidos o diuréticos.
En el examen físico de ingreso se observó leve proptosis del globo ocular derecho y debilidad muscular generalizada, sumados a dolor intenso en la movilización tanto activa como pasiva. Peso: 80 kg, altura: 176 cm. El laboratorio reveló hipofosfatemia persistente de 1.2-1.5-1 mg/dl (VN: 2.7-4.5 mg/dl), las determinaciones de calcio iónico resultaron normales: 1.12-1.13-1.10 mMol/l (VN:1-1.3 mMol/l), fosfatasa alcalina:807-746-605 UI/l (VN: 98-279 UI/l), fosfatasa alcalina ósea: 779 UI/l (VN:31-95 UI/l), clearence de creatinina: 135 ml/min (VN >75 ml/m/1.73 m2), sodio y potasio normales. El dosaje de paratohormona (PTH) fue de 19.6 pg/ml. (VN: 10- 69 pg/ml) y el de 25-hidroxi-colecalciferol de 15.7 ng/ml (VN: 9-63 ng/ml). El proteinograma electroforético, función tiroidea y antígeno prostático fueron normales. El análisis de orina no mostró proteinuria, glucosuria ni acidosis metabólica. La excreción de fosfato urinario inorgánico fue de 1027 mg/24 h (VN: 300-900mg/24 h), calciuria de 105 mg/24h (VN: 80-300 mg/24h), creatininuria: 1755 mg/24 h (VN: 800-2000 mg/24h), la reabsorción tubular de fosfato fue del 70% (VN: 85-95%).
El examen radiológico reveló osteopenia generalizada. En la tomografía (TAC) de tórax-abdomen y pelvis no se hallaron anormalidades. La TAC de macizo cráneo facial y órbitas con contraste endovenoso mostró una imagen de aspecto heterogéneo en seno maxilar derecho y cavidad nasal que reforzaba con contraste, con seno esfenoidal y celdillas etmoidales parcialmente ocupadas. Asimismo evidenció disrupción de la pared interna de la órbita, invasión de la base de cráneo, compromiso lítico de los cornetes y paladar duro horadado (Fig. 1). La tomografía por emisión de positrones (PET/TC con F18-Na) reveló múltiples áreas hipercaptantes esqueléticas, especialmente en esqueleto axial y particularmente en la columna dorsal, sin correlación con la tomografía computarizada. La lesión expansiva en fosa nasal y etmoides no acumulaba el radiotrazador (Fig. 2).
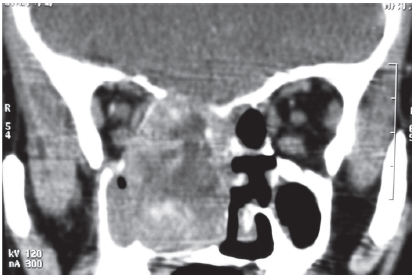
Fig. 1.- TAC de macizo cráneo facial y órbitas con contraste endovenoso: imagen de aspecto heterogéneo en seno maxilar derecho y cavidad nasal que refuerza con contraste y compromete las estructuras óseas adyacentes.

Fig. 2.- PET/TC con F18-Na: múltiples áreas hipercaptantes esqueléticas especialmente en la región axial sin correlación con la tomografía computarizada. La lesión expansiva que compromete la fosa nasal y el etmoides del lado derecho no acumula el radiotrazador.
Se realizó biopsia de tumor rinosinusal cuyo resultado histológico fue de hemangiopericitoma. Posteriormente se procedió a la exéresis tumoral, comprobándose en el laboratorio normalización de los parámetros alterados. Se tomaron muestras de suero del paciente para la determinación del factor de crecimiento fibroblástico 23 (FGF 23) antes, durante y después de la cirugía. Los resultados expresados en pg/ml para un rango normal de 10 a 50 pg/ml fueron los siguientes: antes de la cirugía 129 pg/ml, durante la cirugía 80 pg/ml, una hora después de la cirugía 33 pg/ml y a las dos horas de la cirugía 21 pg/ml.
El paciente presentó en el postoperatorio una neumonía asociada al respirador, evolucionando desfavorablemente con shock séptico con disfunción multiorgánica y falleció a los 30 días de la cirugía.
Discusión
La osteomalacia inducida por tumor es un síndrome causado principalmente por tumores mesenquimatosos secretores de factores fosfatúricos que producen pérdida renal de fosfato y en consecuencia hipofosfatemia1-4. La OIT es una enfermedad adquirida infrecuente del metabolismo óseo, de difícil diagnóstico que se puede presentar a cualquier edad con similar prevalencia en hombres y mujeres6. Están descriptas formas hereditarias de hipofosfatemia ligada al cromosoma X y las hipofosfatemias autosómica dominante y recesiva1, 2.
El desarrollo insidioso de dolor óseo progresivo y la debilidad muscular son características bien conocidas de la osteomalacia, pero que también pueden obedecer a otras causas2. Los síntomas y signos clínicos de la OIT como síndrome paraneoplásico son inespecíficos y pueden conducir a errores diagnósticos con patología osteoarticular o desórdenes neurológicos2. En nuestro caso tanto la presentación clínica como la evaluación diagnóstica inicial fueron adjudicadas a un proceso tumoral con metástasis de tumor primario desconocido. A los tres años del inicio del cuadro clínico consultó a nuestra institución. El examen físico exhaustivo reveló la presencia de una leve protosis del ojo derecho. Este hallazgo motivó la solicitud de estudios por imágenes que sumados a las alteraciones bioquímicas encontradas en el laboratorio nos aproximó al diagnóstico.
Hasta la fecha, más de 160 casos de OIT han sido notificados según la literatura revisada. Los tipos más comunes suelen ser mesenquimatosos o mixtos del tejido conectivo, principalmente con localización en la cabeza y el cuello como el hemangiopericitoma2, 7. Es infrecuente que tumores histológicamente malignos o metástasis a distancia sean el origen de este cuadro2. En su mayoría son de naturaleza benigna, muy vascularizados, de lento crecimiento, y debido a su pequeño tamaño son tumores difíciles de localizar2, 7. Está descripto que cantidades microscópicas de tumor son capaces de causar este síndrome12. Otros tipos de tumores secretores de factores fosfatúricos incluyen fibromas, condrosarcomas, histiocitomas, neuroblastomas, cáncer de próstata y algunos tipos de carcinomas1, 2.
Los marcadores bioquímicos de la OIT son la hipofosfatemia, disminución de la reabsorción tubular renal de fosfato, aumento de la fosfatasa alcalina sérica con la presencia de niveles normales de calcio, 25 hidroxi-vitamina D3 y de PTH. Los niveles en sangre de 1.25 dihidroxi-vitamina D3 son normales a bajos1, 2. Los valores de laboratorio en nuestro paciente fueron coincidentes a los casos descriptos en la literatura.
No se conoce el mecanismo exacto por el cual el tumor induce estos cambios bioquímicos. Las similitudes clínicas entre osteomalacia oncogénica y la condición hereditaria ligada al cromosoma X, y la evidencia de la participación de un factor humoral, han dado lugar a plantear que mecanismos similares podrían estar involucrados en las dos condiciones7. En los pacientes estudiados con osteomalacia familiar fueron identificados factores reguladores de la homeostasis del fosfato llamados "fosfatoninas" [proteína 4 relacionada (sFRP4), fosfoglicoproteína de la matriz extracelular (MEPE), factor de crecimiento fibroblástico 7 (FGF7), y sobre todo el factor de crecimiento fibroblástico 23 (FGF23)]6. Estos factores son producidos por el tejido tumoral y alteran la homeostasis del fosfato en el riñón y el hueso disminuyendo la reabsorción de fósforo al alterar los transportes sodio dependientes5, 6. Además, inhiben la síntesis de vitamina D3 que conduce a una disminución de la absorción intestinal de fosfato5, 8 . En nuestro paciente se pudieron obtener las determinaciones de FGF 23 antes, durante y después de la cirugía, comprobándose la normalización de su valor luego de la resección del tumor y apoyando su rol fisiopatogénico en esta entidad.
El gen responsable en la hipofosfatemia ligado al cromosoma X fue identificado en el cromosoma Xp 22. 115. Un gran número de mutaciones en este gen pueden causar la enfermedad pero todavía no se encuentra una correlación lineal entre genotipo y fenotipo.
El principal desafío radiológico en la osteomalacia oncogénica es la detección del tumor causante, ya que el diagnóstico no puede ser confirmado hasta que el tumor es detectado y extirpado9. Constituye una tarea difícil, ya que el tumor suele ser pequeño y localizarse a nivel del tejido blando y esquelético, y habitualmente pasa desapercibido durante un período considerable de tiempo que oscila entre 5 meses y 16 años con una media de 5 años entre el inicio de los síntomas y la extirpación quirúrgica10, 11. La mayoría de los tumores mesenquimáticos se encuentran en las extremidades y la cabeza, y suelen confundirse con lesiones benignas9, 12. En nuestro paciente se ubicó en el seno maxilar y el tiempo transcurrido desde el inicio de los síntomas hasta su localización, diagnóstico y posterior tratamiento fue de 3 años.
En la búsqueda y detección del tumor se han empleado diversos estudios de imágenes que incluyen: radiografías simples, centellograma óseo, tomografías y resonancia magnética (RM). Recientemente la incorporación del PET/TC ha mostrado ser una técnica prometedora en la localización de tumores pequeños.
Las radiografías simples detectarán en la mayoría de los casos osteopenia significativa, así como zonas de fracturas, pero con frecuencia no logran detectar un tumor asociado11, 12. En las radiografías de nuestro caso también se encontraron los mismos hallazgos descriptos.
La osteomalacia oncogénica puede confundirse con lesiones óseas metastásicas13. El patrón de captación en la centellografía ósea en la OIT y la osteomalacia secundaria a otras causas (por ejemplo, malabsorción, insuficiencia renal, deficiencia de vitamina D, medicamentos, etc.) son casi indistinguibles . Por lo tanto, mediante este estudio no puede ser valorado el comportamiento del tumor. El centellograma óseo corporal total de nuestro paciente fue informado con patrón compatible con secundarismo. La interpretación de metástasis ósea debe leerse con cautela y solo con el conocimiento de que el tumor es maligno y no hay causas metabólicas que permitan explicar los resultados13.
Debido a que diferentes estudios in vitro han revelado que muchos tumores mesenquimáticos expresan receptores de somatostatina, la gamagrafía con octreótide marcado con indium 11 (SPECT/ TC ) ha demostrado ser de valor en la detección y localización de tumores inductores de osteomalacia9, 11.
En ausencia de resultados positivos en las imágenes estándar, un rastreo esquelético con RM es un complemento muy valioso para el estudio diagnóstico radiográfico de osteomalacia oncogénica. Esta valoración incluye: imágenes de cráneo, tórax, abdomen y de la zona proximal apendicular del esqueleto11.
El PET/TC ha mostrado ser de utilidad en el diagnóstico y seguimiento de los casos con OIT, sobre todo en los tumores muy vascularizados como es el caso del hemangiopericitoma10. Otro valor diagnóstico del PET/TC reside en confirmar y localizar los tumores recidivantes debido que los cambios en el postoperatorio pueden generar confusión en la RM11. En nuestro caso, a causa de la demora en el diagnóstico, el tumor alcanzó un tamaño considerable para ser fácilmente localizado por estudios tomográficos y el PET/TC complementó la evaluación diagnóstica.
Esta entidad no tiene tratamiento médico satisfactorio. Se postulan análogos de la somatostatina y suplementos de fósforo y calcitriol1, 4. La eficacia de octreótide como análogo de la somatostatina en el tratamiento de la OIT sigue siendo controvertida14. El tratamiento médico con vitamina D y suplementos orales de fosfato tiene una respuesta variable13. Actualmente se encuentran en desarrollo anticuerpos anti FGF 23 para uso en humanos1.
Una vez que el tumor se ha diagnosticado, se debe intentar la resección quirúrgica13. La resección de tumor, sobre todo si es benigna, puede conducir a la curación permanente de la osteomalacia5, 6. Incluso la resección parcial del tumor puede dar lugar a mejoría de los síntomas. Los tumores malignos son más resistentes a la resección quirúrgica13.
En nuestro caso, la masa tumoral fue extirpada quirúrgicamente y el estudio histopatológico mostró un hemangiopericitoma. Después de la operación, el fosfato sérico regresó a la normalidad y se mantuvo dentro del límite normal hasta el fallecimiento del paciente.
Agradecimientos: Agradecemos a la Dra. Beatriz Olivera del servicio de Osteopatías del Hospital de Clínicas y a la Dra. Suzanne Jan de Beur del Johns Hopkins Hospital por su colaboración en el procesamiento de las muestras y datos.
Conflicto de interés: No existen conflictos financieros o personales que hayan influenciado este trabajo.
1. Bergwitz C, Collins MT, Kamath RS, Rosenberg AE. Case 33-2011: A 56-year-old man with hypophosphatemia. N Engl J Med 2001; 365: 1625-35. [ Links ]
2. Radaideh AR, Jaradat D, Abu-Kalaf MM, Nusier MK. Resolution of severe oncogenic hypophosphatemic osteomalacia after resection of a deeply located soft-tissue tumour. Curr Oncol 2009; 16: 87-90. [ Links ]
3. Jan De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barbeiro D. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res 2002; 17: 1102-10. [ Links ]
4. Seufert J, Ebert K, Müller J, Eulert J, Hendrich C. Octeotride therapy for tumor-induced osteomalacia. N Engl J Med 2001; 345: 1883-8. [ Links ]
5. Geller JL, Khosravi A, Kelly MH, Riminucci M, Adms JS. Cinacalcet in the management of tumor -induced osteomalacia. J Bone Miner Res 2007; 22: 931-7. [ Links ]
6. Hesse E, Moessinger E, Rosenthal H, Laenger F, Brabant G. Oncogenic osteomalacia exact tumor localization by co- Registration of positron emission and computed tomografhy. J Bone Miner Res 2007; 22: 158-62. [ Links ]
7. Carpenter TO. Oncogenic osteomalacia- A complex dance of factors. N Engl J Med 2003; 348: 1705-8. [ Links ]
8. Cotant CL, Rao PS. Elevated fibroblast growth factor 23 in a patient with metastatic prostate cancer and hypophosphatemia. Am J Kidney Dis 2007; 50: 1033-6. [ Links ]
9. Gershinsky M, Croitoru S, Dickstein G, Bardicef O, Gelman R. Imaging of oncogenic osteomalacia. IMAJ 2007; 9: 566-7. [ Links ]
10. Khadgawat R, Singh Y, Kansara S, Tandon N, Bal C. PET/CT localization of a scapular haemangiopericytoma with tumour-induced osteomalacia. Singapore Med J 2009; 50: e55-e57. [ Links ]
11. Chua SC, O`Connor SR, Wong WL, Ganatra RH. Solitary plasmacytoma of bone with oncogenic osteomalacia: recurrence of tumour confirmed by PET/CT. A case report with a review of the radiological literature. Br J Radiol 2008; 81: e110-e114. [ Links ]
12. Weiss D, Bar RS, Weidner N, Wener M, Lee F. Oncogenic osteomalacia: strange tumours in strange places. Postgrad Med J 1985; 61: 349-55. [ Links ]
13. Lee HK, Wen Sung W, Salodnik P, Shimshi M. Bone scan in tumor-induced osteomalacia. J Nucl Med 1995; 36: 247-9. [ Links ]
14. Nasu T, Kurisu S, Matsuno S, Tatsumi K, Kakimoto T. Tumor-induced hypophosphatemic osteomalacia diagnosed by the combinatory procedures of magnetic resonance imaging and venous sampling for FGF 23. Inter Med 2008; 47: 957-96. [ Links ]
15. Francis F, Hennig S, Korn B, et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 1995; 11: 130-6. [ Links ]
Recibido: 9-I-2012
Aceptado: 14-VI-2012

