










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680
Medicina (B. Aires) vol.73 no.2 Ciudad Autónoma de Buenos Aires Apr. 2013
ARTÍCULO ORIGINAL
Vasculitis asociadas a anticuerpos anti-citoplasma de neutrófilos. Clínica y tratamiento
María Virginia Paolini, Juan Pablo Ruffino, Diego S. Fernández Romero
Unidad de Inmunología e Histocompatibilidad, Hospital Dr. Carlos G. Durand, Buenos Aires
Dirección Postal: Dra. María V. Paolini, Mendoza 2977, 1428 Buenos Aires, Argentina
Fax: (54 -11) 4781-6446 e-mail: virpaolini@gmail.com
Resumen
Las vasculitis asociadas a anticuerpos anti-citoplasma de neutrófilos (ANCA) comprenden a un grupo de enfermedades caracterizadas por la inflamación de la pared de pequeños vasos. Analizamos las características epidemiológicas y clínicas en una serie de 47 pacientes: 23 (49%) granulomatosis de Wegener (GW), 15 (32%) poliangeítis microscópica (PAM) y nueve (19%) vasculitis limitada al riñón (VLR). La edad media al inicio de los síntomas fue de 50.7 ± 14.9 años. La manifestación clínica más frecuente fue el compromiso renal en 41 (87%) pacientes, seguido por el pulmonar en 26 (55%) y el otorrinolaringológico en 17 (36%). En 26 (55%) se asoció compromiso renal y pulmonar. La forma clínica más frecuente fue la generalizada en 23 (49%), seguida por la grave en 18 (38%). El 89% presentaron determinaciones de ANCA positivas. Cuatro (8%) no recibieron tratamiento inmunosupresor de inicio. De los 43 que recibieron tratamiento de inicio, 29 (67%) tuvieron remisión completa, con un tiempo de remisión promedio de 35.3 meses. Once (26%) presentaron recaídas, diez (91%) recaídas mayores y uno (9%) menor. Doce (28%) fallecieron, siete en forma temprana y cinco durante la evolución de la enfermedad. Quince (31%) evolucionaron a insuficiencia renal crónica. Los 26 pacientes en seguimiento tuvieron respuesta al tratamiento y 20 (77%) de ellos estaban en remisión al finalizar el estudio. Las vasculitis asociadas a ANCA continúan siendo enfermedades de alta morbilidad y mortalidad, a pesar de las mejorías logradas con los tratamientos inmunosupresores.
Palabras clave: Vasculitis asociadas a ANCA; Granulomatosis de Wegener; Poliangeítis microscópica; Vasculitis limitada al riñón
Abstract
Anti-neutrophil cytoplasmic antibody-associated vasculitis. Clinical aspects and treatment. Anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis, comprise a group of diseases characterized by inflammation of the wall of small vessels. We analyzed epidemiological and clinical characteristics in a series of 47 patients, 23 (49%) with Wegener granulomatosis (WG), 15 (32%) with microscopic polyangiitis (MPA) and nine (19%) with renal limited vasculitis (RLV). The mean age at onset of symptoms was 50.7 ± 14.9 years. The most frequent clinical manifestation was renal involvement in 41 (87%), followed by pulmonary manifestations in 26 (55%) and ENT involvement in 17 (36%). In 26 (55%) it presented with simultaneous pulmonary and renal involvement. The most frequent clinical category was the generalized form in 23 (49%), followed by the severe form in 18 (38%). Eighty nine percent of patients had positive ANCA test. Four (8%) received no immunosuppressive treatment. Of the 43 patients who were treated, 29 (67%) achieved complete remission with an average length of remission of 35.3 months. Eleven (26%) had a relapse, ten (91%) had a major relapse and one had a minor relapse. Twelve (28%) patients died, seven died early and five late during the course of the disease. Fifteen (31%) progressed to chronic renal failure. All 26 patients in follow-up had response to treatment and 20 (77%) were in remission at the end of the study. Despite the improvements achieved with immunosuppressive treatments, morbidity and mortality rates in ANCA-associated vasculitis remain high.
Key words: ANCA-associated vasculitis; Wegener granulomatosis; Microscopic polyangiitis; Renal limited vasculitis
Las vasculitis asociadas a anticuerpos anti-citoplasma de neutrófilos (ANCA) comprenden a la granulomatosis de Wegener (GW), la poliangeítis microscópica (PAM), el síndrome de Churg-Strauss (SChS) y la glomerulonefritis necrotizante pauci-inmune o vasculitis limitada al riñón (VLR)1. Se caracterizan por la inflamación y necrosis de la pared vascular de pequeños vasos y todas comparten, con una prevalencia variable, la presencia de ANCA. Aproximadamente el 90% de los pacientes tienen autoanticuerpos contra la proteína mieloperoxidasa (MPO-ANCA) o contra la enzima proteinasa-3 (PR3-ANCA)2.
Este grupo de entidades se considera la forma más común de vasculitis sistémica primaria de pequeños vasos en adultos3, 4. La incidencia anual global de las vasculitis asociadas a ANCA, con tendencia en aumento en los últimos años, es de 20 casos por 1 000 000 de habitantes, siendo de entre 2.1 a 15 en GW, 2.1 a 17.5 en PAM y 0.5 a 3.1 en SChS, mostrando variaciones geográficas e inter-étnicas. El pico de incidencia de las mismas se observa en el grupo etario de 55 a 74 años5.
Las vasculitis de pequeños vasos comparten características patogénicas y patológicas y pueden tener formas clínicas similares6-8. Las manifestaciones clínicas son variadas, pudiendo limitarse solo al riñón o comprometer la vía aérea superior, los pulmones, la piel y otros órganos en diferentes combinaciones. El compromiso renal ocurre en el 70% de los pacientes y se suele manifestar como una glomerulonefritis rápidamente progresiva. El compromiso pulmonar se observa en aproximadamente la mitad de los pacientes y puede variar desde cuadros clínicos con infiltrados pulmonares o nódulos hasta formas severas como la hemorragia pulmonar2, 7, 8.
El tratamiento se basa principalmente en el uso de un esquema de inducción con glucocorticoides y ciclofosfamida9, 10. A pesar que con el uso de agentes alquilantes la tasa de mortalidad se redujo en un 62%, lográndose la remisión en aproximadamente el 70-90% de los pacientes, la mortalidad en el primer año excede el 15%, el 50-66% de los pacientes evolucionan a insuficiencia renal crónica (IRC) terminal y la recurrencia de la enfermedad es del orden del 30 al 50%2, 9-11. Es en este marco en el que surge la necesidad de nuevas terapias para mejorar la efectividad y disminuir la toxicidad de los tratamientos actuales. La plasmaféresis asociada a la inmunosupresión para casos de gravedad demostró mejorar la sobrevida renal y el uso de agentes biológicos como alternativa a las terapias convencionales parece ser una futura opción terapéutica12, 13.
El objetivo de este estudio fue describir nuestra experiencia en una población de 47 pacientes con diagnóstico de vasculitis asociada a ANCA, sus características clínicas, epidemiológicas, los tratamientos instaurados y la respuesta a los mismos.
Materiales y métodos
El estudio incluyó a todos los pacientes con diagnóstico de vasculitis asociadas a ANCA asistidos en la Unidad de Inmunología del Hospital Dr. Carlos G. Durand de la Ciudad de Buenos Aires, en el período comprendido entre noviembre de 1997 y septiembre de 2011. El diagnóstico de vasculitis y su clasificación se realizó en base a las características clínicas, hallazgos histopatológicos y/o presencia de marcadores específicos según los criterios diagnósticos del American College of Rheumatology, la Conferencia de Consenso de Chapel Hill para vasculitis sistémicas primarias asociadas a ANCA y las recomendaciones del Grupo EULAR (European League Against Rheumatism) para conducir estudios o ensayos clínicos en vasculitis sistémicas primarias1, 14-16.
Se analizaron las historias clínicas de cada paciente consignando las características epidemiológicas, las manifestaciones clínicas al inicio y durante la evolución, la extensión y la clasificación de la enfermedad, el tratamiento instaurado y la respuesta al mismo, y el daño provocado por la enfermedad. Se analizaron los datos de laboratorio y anatomopatológicos
La estadificación de enfermedad en cada paciente se estableció de acuerdo a la clasificación del Grupo EUVAS (European Vasculitis Study Group) en localizada, sistémica temprana, generalizada, grave y refractaria17.
Se denominaron refractarios a aquellos pacientes que no presentaran cambios en la actividad o aumentaran la misma luego de cuatro semanas de tratamiento convencional con glucocorticoides y ciclofosfamida para las formas agudas, o a los que no tuvieron al menos 50% de la disminución de la actividad luego de seis semanas; o enfermedad crónica persistente luego de 12 semanas de tratamiento convencional16, 17.
Se consideró respuesta al tratamiento a una reducción mayor o igual al 50% en la actividad de la enfermedad, evaluada utilizando el Birmingham Vasculitis Activity Score (BVAS) y BVAS/GW18, 19. Se definió como remisión a la ausencia completa de actividad de la enfermedad, incluyendo no solo las manifestaciones vasculíticas sino también a las granulomatosas, por al menos tres meses con una dosis máxima de glucocorticoides de 10 mg/día de prednisona o su equivalente. Se consideró recaída a la recurrencia o nueva aparición de actividad atribuible a inflamación activa, y se las diferenció en mayores cuando comprometían a un órgano vital o amenazaban la vida, y menores cuando no reunían los requisitos anteriores16. Se definió muerte temprana a la ocurrida durante los primeros seis meses de la enfermedad11.
El daño permanente ocasionado por la enfermedad se consideró como la evolución a insuficiencia renal terminal, enfermedad pulmonar crónica, hipoacusia, deformidad en golpe de sable, ceguera o muerte9.
En aquellos pacientes en los cuales el tiempo desde la última consulta fue mayor a seis meses se realizó un llamado telefónico y se obtuvieron datos respecto a la evolución de la enfermedad y controles médicos realizados. Consideramos paciente en seguimiento a todos aquellos que fueron evaluados en los últimos dos meses previos a la finalización de la recolección de datos o que se encuentran en contacto a distancia. Se consideró tiempo de seguimiento al tiempo transcurrido desde la fecha de primera consulta hasta la de la última consulta, contacto telefónico u óbito.
En todos los casos se realizaron análisis de laboratorio con determinación de ANCA por inmunofluorescencia indirecta (IFI), de anticuerpos anti-PR3, anti-MPO y anti-membrana basal glomerular (AMBG) por enzimoinmunoanálisis (ELISA), anticuerpos anti-nucleares (ANA) por IFI utilizando como sustrato células Hep-2, determinación de los componentes C3 y C4 del complemento por inmunodifusión radial o nefelometría, y crioglobulinas.
Resultados
El estudio incluyó a 47 pacientes con diagnóstico de vasculitis asociadas a ANCA, 29 mujeres (62%) y 18 varones (38%), con una edad promedio en el momento de la última consulta de 54.1 ± 15.9 años (rango 20 a 83). En 23 (49%) se diagnosticó GW, 11 mujeres y 12 varones, con una edad promedio en el momento de la última consulta de 55.1 ± 16.1 años (rango 26 a 82). En 15 (32%) se diagnosticó PAM, 12 mujeres y tres varones, con una edad promedio en el momento de la última consulta de 57.9 ± 16.1 años (rango 31 a 83). En nueve (19%) se diagnosticó VLR, seis mujeres y tres varones, con una edad promedio en el momento de la última consulta de 46.9 ± 15.1 años (rango 20 a 62). Al finalizar el estudio, 26 (55%) continuaban en seguimiento, en nueve (19%) se perdió el seguimiento y 12 (26%) habían fallecido. El tiempo promedio de seguimiento fue de 35.2 ± 38.9 meses (rango 1 a 150) (Tabla 1).
TABLA 1.- Vasculitis asociada a ANCA: pacientes, características, demora diagnóstica y tiempo de seguimiento

(1)Expresado en número de pacientes y porcentaje respecto al total; (2)Edad expresada en años: promedio ± DE (rango); (3)Tiempo entre inicio de síntomas y diagnóstico expresada en meses: promedio ± DE (rango). (4)Tiempo de seguimiento expresado en meses: promedio ± DE (rango). GW: granulomatosis de Wegener; PAM: panarteritis microscópica, VLR: vasculitis limitada al riñón.
La edad de inicio de síntomas fue de 50.6 ± 14.9 años (rango 20 a 77). La edad promedio en el momento del diagnóstico fue de 51 ± 15 años (rango 20 a 77). El tiempo promedio entre el inicio de los síntomas y el momento del diagnóstico fue de 6.1 ± 11.1 meses (rango 1 a 60) (Tabla 1). En 31 (66%) pacientes el tiempo entre el inicio de los síntomas y el diagnóstico fue de 0-3 meses, en cuatro (8%) de 3-6 meses, en siete (15%) de 6-12 meses y en cinco (11%) de más de 12 meses.
Las formas clínicas de inicio más frecuentes fueron: el compromiso renal en 38 (81%) pacientes, seguido por el pulmonar en 17 (36%), el otorrinolaringológico (ORL) en 11 (23%), el oftálmico en seis (13%), cutáneo en tres (6%), del sistema nervioso periférico (SNP) en tres, del sistema nervioso central (SNC) en uno (2%), y del tejido mamario en otro. Inicialmente, 17 (36%) asociaron el compromiso renal al pulmonar (RP) y seis (13%) presentaron simultáneamente compromiso renal, pulmonar y otorrinolaringológico (RPO). Treinta y siete (79%) presentaron insuficiencia renal de inicio, de los cuales 26 (55%) requirieron hemodiálisis (Tabla 2).
TABLA 2.- Manifestaciones clínicas al inicio de la enfermedad y durante el seguimiento

Seg.: Seguimiento, ORL: otorrinolaringológico, SNP: sistema nervioso periférico, SNC: sistema nervioso central, RP: renal-pulmonar, SRP: síndrome riñón pulmón, RPO: renal-pulmonar-otorrinolaringológico
Durante la evolución de la enfermedad, 41 (87%) pacientes presentaron compromiso renal, 26 (55%) pulmonar, 17 (36%) otorrinolaringológico, 11 (23%) oftálmico, nueve (19%) cutáneo, siete (15%) del sistema nervioso periférico, cuatro (8%) articular, tres (6%) del sistema nervioso central, uno (2%) gastrointestinal y otro de tejido mamario. Veintiséis (55%) asociaron el compromiso RP y 11 de ellos presentaron compromiso RPO (Tabla 2).
De los 26 con afectación pulmonar, 16 (61%) presentaron hemorragia alveolar asociada a compromiso renal como síndrome riñón-pulmón (SRP). De ellos, nueve (2 PAM y 7 GW) presentaron SRP como forma de inicio, y siete durante la evolución de la enfermedad.
Según la clasificación del Grupo EUVAS14, durante la evolución de la enfermedad, 23 pacientes (49%) presentaron formas generalizadas (G), 18 (38%) graves (S), cinco (11%) limitadas (L), uno (2%) sistémica temprana (ST) y cuatro (8%) refractarias (R). Las formas refractarias comprendieron a tres pacientes con GW, uno con afectación localizada y dos con compromiso generalizado, y uno con PAM generalizada (Tabla 3).
TABLA 3.- Estadificación de la enfermedad según clasificación del Grupo EUVAS14

GW: granulomatosis de Wegener, PAM: poliangeítis microscópica, VLR vasculitis limitada al riñón
De los 47 pacientes, 42 (89%) presentaron ANCA positivo. De ellos, 21 (50%) correspondieron a un patrón citoplasmático (ANCA-c), todos con GW, y 21 (50%) correspondieron a un patrón perinuclear (ANCA-p): 15 con diagnóstico de PAM, cinco VLR y uno GW. En cinco (4 VLR y 1 GW limitada) la determinación de ANCA fue negativa. La especificidad de ANCA correspondió a la presencia de anticuerpos anti-PR3 en todos los casos con ANCA-c positivo y en un caso de ANCA-p positivo (en un paciente con GW), y a la presencia de anticuerpos anti-MPO en 20 casos de ANCA-p positivos (en pacientes con diagnóstico de PAM y VLR). El resto de los estudios inmunológicos realizados (ANA, AMBG, Crioglobulinas, C3, C4) no arrojaron datos significativos, descartándose otras entidades con formas de presentación similar.
En 38 (81%) pacientes se realizaron biopsias al inicio de la enfermedad; en todas ellas se informaron hallazgos compatibles con vasculitis. Treinta y tres correspondieron a biopsias renales, dos de senos paranasales, dos de tejido orbitario y una de nervio periférico y cutánea simultánea. En cuatro pacientes en los que se realizó biopsia renal también se biopsiaron otros tejidos (pulmonar, cutáneo y mamario).
De los 47 pacientes, cuatro (8%), con IRC terminal y requerimiento de diálisis, no recibieron tratamiento inmunosupresor de inicio. De estos cuatro, tres tenían diagnóstico de VLR: uno falleció a los días de realizado el diagnóstico; el segundo discontinuó el seguimiento; y el último no recibió tratamiento por presentar esclerosis renal sin manifestaciones extrarrenales, continuando en seguimiento. El restante, con PAM, inició la enfermedad con el compromiso renal y el diagnóstico se realizó en forma retrospectiva luego de 13 meses por compromiso pulmonar; actualmente continúa en seguimiento y se encuentra en remisión a pesar de no haber recibido tratamiento durante la evolución de su enfermedad.
De los 43 que recibieron tratamiento de inicio, 32 (74%) fueron tratados con glucocorticoides (GC) y ciclofosfamida (CF), 6 (14%) con GC como monoterapia, cuatro (9%) con GC y metotrexato (MTX) y uno (2%) con GC y azatioprina (AZA). (Tabla 4).
TABLA 4.- Pacientes con vasculitis asociada a ANCA. Tratamiento inicial
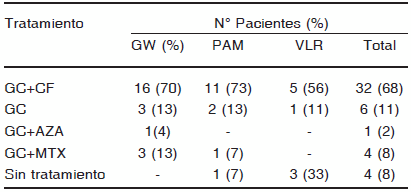
GW: granulomatosis de Wegener, PAM: poliangeítis microscópica, VLR vasculitis limitada al riñón, GC: glucocoticoides, CF: ciclofosfamida, AZA: azatioprina, MTX: metotrexate
De los 32 que fueron tratados con CF y GC, en 30 la administración de CF se realizó en forma de pulsos mensuales endovenosos (EV), y en dos, por vía oral (VO). De ellos, 15 presentaron formas generalizadas, 16 graves y solo uno tuvo una forma localizada. De los 30 que recibieron GC y CF EV, nueve (9%) realizaron plasmaféresis simultáneamente (8 por compromiso RP, y uno por compromiso renal y oftalmológico).
Seis (13%) recibieron solo GC: tres con diagnóstico de GW, dos con PAM y uno con VLR. De estos seis, cinco (83%) presentaron formas generalizadas de inicio y uno compromiso localizado. De los que presentaron formas generalizadas, tres debutaron con compromiso renal, dos de ellos con IR y requerimiento de diálisis sin actividad extrarrenal, y dos con SRP. Cuatro fueron tratados con MTX y GC; tres de ellos presentaban GW localizadas y uno PAM sistémica temprana. Uno con GW localizada de inicio fue tratado durante siete meses con GC y AZA por fibrosis pulmonar; durante el tratamiento presentó una recaída pulmonar con compromiso renal y murió al mes de haber recibido CF EV.
De los 30 tratados con CF EV y GC, seis (20%) fallecieron durante el período de inducción, cinco (17%) se encuentran realizando el tratamiento de inducción con respuesta, de dos (6.5%) se desconoce la evolución posterior inmediata al inicio del tratamiento y dos (6.5%) realizaron tratamiento por menos de seis meses. En los 15 (50%) restantes, que completaron los seis meses de tratamiento de inducción, se realizó un descenso gradual de GC; en 11 (73%) de ellos se continuó el tratamiento de mantenimiento con pulsos de CF trimestrales y en cuatro (27%) con azatioprina por vía oral.
De los 32 tratados con GC-CF, 24 (75%) presentaron respuesta al tratamiento, 19 (59%) con remisión completa, con una duración promedio del tratamiento de 19.7 meses (rango 2 a 54), y un tiempo promedio de remisión de 42.6 meses (rango 6 a 143). Siete (4 GW y 3 PAM) de los 19 con remisión completa (37%) presentaron recaídas mayores comprometiendo el riñón o el pulmón. Seis habían realizado tratamiento por menos de dos años; cuatro de estos con suspensión del tratamiento por evolución a IRC y ausencia de actividad extrarrenal, uno con suspensión de la terapia por dosis acumulada de CF y otro por recaídas durante el mantenimiento.
Todos los que recibieron sólo GC tuvieron respuesta inicial con remisión completa, con un tiempo promedio de remisión de 32.5 meses (rango 6 a 57). Tres (50%) presentaron recaídas: dos PAM con recaídas mayores comprometiendo el pulmón y una GW con una recaída menor con compromiso otorrinolaringológico y oftalmológico. Ninguno de ellos realizó tratamiento por más de un año; la duración de tratamiento promedio fue de 7.6 meses (rango 2 a 12).
De los cuatro tratados con GC y MTX, tres, actualmente en tratamiento, se encuentran en remisión, con un tiempo promedio de remisión de cinco meses (rango 3 a 7). El restante, con GW orbitaria, no respondió a los GC asociados a MTX ni a CF en pulsos mensuales EV, ni a la radioterapia, por lo cual se lo consideró refractario y se lo trato con AZA y rituximab lográndose respuesta por más de 12 meses.
En forma global, considerando a los 43 que recibieron tratamiento, 33 (77%) presentaron respuesta y 29 (67%) remisión completa, con un tiempo de remisión promedio de 35.3 meses (rango 2 a 143). Once (26%) presentaron recaídas. En 10 (91%) fueron recaídas mayores y en un caso, con GW (9%), se trató de una recaída menor, con compromiso ORL y orbitario (Tabla 5).
TABLA 5.- Tratamiento de pacientes con recaídas de la enfermedad

GW: granulomatosis de Wegener, PAM: poliangeítis microscópica, VLR vasculitis limitada al riñón, GC: glucorticoides, CF: Ciclofosfamida, AZA: azatioprina, RTX: rituximab, MTX: metotrexate, PL: plasmaferesis, Enf.: enfermedad, estadif: estadificación, Trat.: Tratamiento, D de trat.: duración de tratamiento
P: pacientes, G: formas generalizadas, S: formas graves Re: formas refractarias R: renal, P: Pulmonar, ORL otorrinolaringológica, SNC: sistema nervioso central, SNP: sistema nervioso periférico, OFT oftalmológico, C: cutáneo, GI gastrointestinal. IRC insuficiencia renal crónica, DAV disminución de agudeza visual. desc: desconocido, RESP: respuesta.
De los 11 tratados por sus recaídas, dos discontinuaron el seguimiento, cuatro fallecieron debido a la recaída de la enfermedad, tres presentaron remisión completa y dos se comportaron como refractarios; en uno de estos casos se hizo necesario agregar un agente biológico a la inmunosupresión para lograr respuesta y el otro falleció por una causa no relacionada con actividad de la enfermedad.
Del total de los tratados cuatro (9%) fueron refractarios, tres GW y una PAM. Tres de ellos luego de una recaída, a pesar de haber logrado la remisión inicial, y el restante con actividad persistente desde el inicio. En dos se pudo agregar al cambio de inmunosupresor rituximab obteniéndose respuestas mayores al año. Los dos restantes evolucionaron, uno con actividad persistente a pesar del cambio de inmunosupresión y el otro con recaidas intratratamiento (Tabla 6).
TABLA 6.- Tratamiento de pacientes con enfermedad refractaria

GW: granulomatosis de Wegener, PAM: panarteritis microscópica, VLR vasculitis limitada al riñón, Enf.: enfermedad, estadif.: estadificación, trat. Tratamiento, act. Persistente: actividad persistente, GC: glucorticoides, CF: ciclofosfamida, AZA: azatioprina, RTX: rituximab, MTX: metotrexate, PL: plasmaferesis, D de trat.: duración de tratamiento. P: pacientes, G: formas generalizadas, S: formas graves Re: formas refractarias R: renal, P: Pulmonar, ORL otorrinolaringológica, SNC: sistema nervioso central, SNP: sistema nervioso periférico, OFT oftalmológico, C: cutáneo, GI gastrointestinal. IRC insuficiencia renal crónica, DAV disminución de agudeza visual. desc: desconocido, RESP: respuesta.
De los 26 en seguimiento, todos tuvieron respuesta al tratamiento al final del estudio, y 20 (77%) alcanzaron la remisión, encontrándose 13 de ellos sin tratamiento y siete en tratamiento.
En relación a las secuelas asociadas a la enfermedad, entre los 41 pacientes con compromiso renal, 15 (37%) evolucionaron a IRC (7 PAM, 4 GW, 4 VLR). De los 17 con GW con compromiso ORL, dos (12%) evolucionaron con deformidad en golpe de sable y otros dos con hipoacusia. De los 11 con GW y afectación oftálmica, cuatro (36%) tuvieron pérdida de visión. De los 26 con PAM y compromiso pulmonar, uno (4%) evolucionó a una afección pulmonar crónica.
Durante el estudio fallecieron 12 (26%): nueve (75%) con GW, dos (17%) con PAM y uno con VLR (8%). Siete (58%) murieron en forma temprana (5 GW, 1 PAM, 1 VLR), todos los cuales tuvieron formas de compromiso grave. Cinco (42%) fallecieron durante el seguimiento, cuatro de ellos por una recaída (2 GW generalizadas, 1 GW grave, 1 PAM generalizada refractaria) y el restante (GW generalizada) por una causa no identificada.
Discusión
Presentamos una serie de 47 pacientes con diagnóstico de vasculitis asociadas a ANCA. La enfermedad más frecuentemente diagnosticada fue la GW, en casi la mitad de los casos, seguida por la PAM y la VLR, coincidiendo con lo descripto en otras series4, 5. La ausencia de pacientes con diagnóstico de SChS en nuestra serie podría deberse a su menor prevalencia o a que éstos son asistidos por otras especialidades debido a su forma de presentación clínica inicial con rinitis, pólipos nasales y asma.
La edad promedio al diagnóstico fue similar a la comunicada por otros autores, excepto en la PAM que fue inferior4, 5. A pesar de que en la bibliografía no se ha podido determinar un claro predominio de sexo, hemos observado una distribución a favor del sexo femenino, a expensas de los pacientes con PAM y VLR, coincidiendo con los resultados de Di Benedetto y col.20. En el subgrupo con GW, al igual que en otros informes, no se observó tal predominio4, 5, 20.
En la mayoría de los casos el diagnóstico se realizó dentro de los tres meses a partir del comienzo de los síntomas, correspondiendo principalmente a pacientes con compromiso renal, pulmonar o la asociación de ambos, y, por ende, con mayor sospecha diagnóstica inicial. Considerando cada enfermedad por separado, el retraso diagnostico fue menor en el grupo con VLR, posiblemente debido a la presentación con formas de compromiso renal de gravedad. Los casos con mayor demora diagnóstica comprendieron a las formas localizadas y formas iniciales atípicas o con insuficiencia renal terminal, sin sospecha de la enfermedad hasta la aparición de una manifestación extrarrenal posterior. Estas presentaciones también se han asociado a más demora diagnóstica en la bibliografia2, 9, 10.
Las formas clínicas de inicio más frecuentes fueron las caracterizadas por compromiso renal, pulmonar, otorrinolaringológico y oftálmico, en forma coincidente con lo informado en la literatura6, 7, 9. La frecuencia de compromiso renal fue levemente superior a la comunicada en otras series, en las cuales no sobrepasa el 80%7. Esto podría explicarse por la inclusión del subgrupo de pacientes con diagnóstico de VLR, y por la alta frecuencia de compromiso renal en el subgrupo con PAM de nuestra serie6, 10. La frecuencia de compromiso renal en la GW fue similar a lo informado previamente6, 9. La insuficiencia renal de inicio se observó en el 79%, requiriendo iniciar hemodiálisis en más de la mitad de los casos. Estas cifras elevadas con respecto a otras series representarían una mayor gravedad en la presentación en nuestro grupo9, 10. La asociación de manifestaciones renales y pulmonares se observó en más de la mitad de los pacientes, con una frecuencia similar a la comunicada en la bibliografía2.
El compromiso pulmonar fue más frecuente en la PAM, con una incidencia elevada en relación al 30-50% mencionada en otras series6, 10, 22. En el subgrupo con GW se observó una menor frecuencia de compromiso pulmonar con respecto al 85-90% descriptos6, 9, 10, 22. La forma de presentación más frecuente fue la hemorragia alveolar, de modo similar a lo observado por otros autores23. Esta manifestación se observó, en algunos casos, como la primera expresión clínica de la enfermedad, siendo esto más frecuente en el subgrupo con GW, mientras que, en otros casos, se presentó durante el curso de la enfermedad, particularmente entre los pacientes con PAM. Es de notar que todos los pacientes de nuestra serie con afectación pulmonar presentaron, además, compromiso renal, no habiéndose observado compromiso pulmonar aislado, en contraste con lo descripto por otros grupos21.
La forma de presentación más frecuente entre los que presentaron compromiso ORL, al igual que en otras series publicadas, fue la rinosinusitis, observada especialmente entre pacientes con GW y, en menor medida, en pacientes con PAM7, 9, 23. La frecuencia de compromiso ORL en el subgrupo con GW fue menor a la esperada6, 9. El compromiso oftálmico se observó sólo en aquellos con GW, predominando las formas de presentación con escleritis/epiescleritis.
La frecuencia de compromiso de sistema nervioso fue levemente inferior respecto a la descripta en la literatura, y predominantemente involucrando al SNP2, 6. Se observó principalmente entre los pacientes con PAM, aunque su incidencia resulta inferior a la comunicada10. En nuestra serie sólo una baja proporción presentó formas de compromiso limitado, correspondiendo a afectación ORL, oftálmica o ambas.
La presencia de ANCA en suero se observó en todos los pacientes con formas de vasculitis con compromiso sistémico, en la mayoría de quienes presentaron formas localizadas de GW y en la mitad de las formas limitadas al riñón, confirmando la utilidad clínica del marcador en el diagnóstico de formas sistémicas y en la GW localizada, y la posible ausencia del mismo en la vasculitis limitada al riñón6, 7, 9, 24.
Con respecto al tratamiento, las tasas de respuesta, remisión y recaídas fueron similares a las comunicadas en otras series2, 9, 11. En la mayoría de los casos, debido a la gravedad inicial del cuadro clínico, se realizó tratamiento de inducción con CF y GC, observándose en este grupo tasas de respuesta y de remisión completa inferiores a los comunicados previamente en la literatura2, 9, 11. Estas diferencias observadas podrían estar relacionadas a una mayor gravedad en la presentación entre los pacientes incluidos en nuestra serie. La tasa de recaídas fue similar a la informada por otros autores2, 9, 11.
La tasa de recaídas para GW es del 50% y para PAM del 30-34% según diferentes series2, 9-11. Comparadas con estos datos, en nuestro grupo fueron menos frecuentes para GW y coincidentes para PAM, siendo las mismas mayores pulmonares o renales. El tratamiento en forma incompleta se puede relacionar con la posibilidad de recaídas ya que casi la totalidad de los que experimentaron recaídas no completaron el tratamiento. En el grupo con VLR no se observaron recaídas considerando que estos casos evolucionaron con el tratamiento hacia una normalización de la función renal, sin recaídas posteriores; o hacia la insuficiencia renal crónica o terminal y/o la muerte.
Sólo en una baja proporción se observaron presentaciones clínicas refractarias. Entre éstas predominaron las formas de GW localizada con compromiso oftalmológico, particularmente orbitario. A pesar de ser formas de progresión lenta sin compromiso de órganos vitales, la actividad persistente en estos casos llevó a secuelas irreversibles como la afectación de la visión, de modo similar a lo descripto por otros autores25, 26.
En la literatura actual existe controversia acerca de la utilidad del tratamiento con el anticuerpo monoclonal anti-CD20 rituximab en las formas granulomatosas27. A pesar de ello, dos pacientes de nuestra serie que se comportaron como refractarios a esquemas de primera línea y fueron luego tratados con este agente, asociado a otros inmunosupresores, presentaron respuesta al tratamiento por un tiempo superior a un año, encontrándose sin actividad al momento de finalizar el estudio.
La mortalidad en nuestra serie fue elevada en comparación con las tasas informadas por otros autores, que varían del 8 al 20%9, 11. Una importante proporción falleció en forma temprana; todos ellos presentaron formas de compromiso grave. Todos los óbitos acontecidos en forma tardía por una causa asociada a la enfermedad, ocurrieron por una recaída mayor.
Dentro de las distintas entidades clínicas consideradas, la GW presentó la mayor gravedad desde el inicio y mayor tasa de mortalidad. Los pacientes con PAM presentaron una mortalidad menor, a pesar de una mayor tasa de recaídas, y fue la entidad que más frecuentemente evolucionó a IRC. Los pacientes con VLR no presentaron una mortalidad elevada ni recaídas, pero una proporción considerable desarrolló IRC. Al comparar la frecuencia de manifestaciones y formas de evolución observadas en nuestra serie con la comunicada previamente en otros trabajos, debe considerarse como posible limitación el menor número de pacientes incluidos.
Las vasculitis asociadas a ANCA continúan siendo enfermedades de alta morbilidad y mortalidad, a pesar de las mejorías logradas con los tratamientos inmunosupresores.
Conflictos de interés: Los autores declaran no tener conflictos de interés.
1. Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994; 37: 187-92. [ Links ]
2. Falk RJ, Nachman PH, Hogan SL, Jennette JC. ANCA glomerulonephritis and vasculitis: a Chapel Hill perspective. Semin Nephrol 2000; 20: 233-43. [ Links ]
3. Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis. Changing incidence or definition. Semin Arthritis Rheum 1995; 25: 28-34. [ Links ]
4. Watts RA, Lane SE, Bentahm G, Scott DG. Epidemiology of systemic vasculitis. A ten-year study in the United Kingdom. Arthritis Rheum 2000; 43: 414-9. [ Links ]
5. Gibelin A, Maldini C, Mahr A. Epidemiology and etiology of wegener granulomatosis, microscopic polyangiitis, churg-strauss syndrome and goodpasture syndrome: vasculitides with frequent lung involvement. Semin Respir Crit Care Med 2011; 32: 264-73. [ Links ]
6. Jennette JC, Falk RS. Small-vessel vasculitis. N Engl J Med 1997; 337: 1512-23. [ Links ]
7. Seo P, Stone JH. The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med 2004; 117: 39-50. [ Links ]
8. Niles JL, Böttinger EP, Saurina GR, et al. The syndrome of lung hemorrhage and nephritis is usually an ANCA-associated condition. Arch Intern Med 1996; 156: 440-5. [ Links ]
9. Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116: 488-98. [ Links ]
10. Guillevin L, Durand-Gaasselin B, Cevallos R, et al. Microscopic polyangiitis. Clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999; 42: 421-30. [ Links ]
11. Nachman PH, Hogan SL, Jennette JC, Falk RJ. Treatment response and relapse in antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol 1996; 7:33-9. [ Links ]
12. Jayne DRW, Gaskin G, Rasmussen N, et al; European Vasculitis Study Group. Randomized trial of plasma exchange or high-dosage metilprenisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 2007; 18: 2180-8. [ Links ]
13. Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA associated vasculitis. N Engl J Med 2010; 363: 221-32. [ Links ]
14. Hunder GG, Arend WP, Bloch DA, et al. The American College of Rheumatology 1990 criteria for classification of vasculitis. Introduction. Arthritis Rheum 1990; 33: 1065-7. [ Links ]
15. Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener´s granulomatosis. Arthritis Rheum 1990, 33: 1101-7. [ Links ]
16. Hellmich B, Flossmann O, Gross WL, et al. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: focus on anti-neutrophil cytoplasm antibody associated vasculitis. Ann Rheum Dis 2007; 66: 605-17. [ Links ]
17. European Community Study Group on Clinical Trials in Systemic Vasculitis. ECSYSVASTRIAL. European therapeutic trials in ANCA associated systemic vasculitis: disease scoring, consensus, regimens and proposed clinical trials. Clin Exp Immunol 1995; 10: 29-34. [ Links ]
18. Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. Quart J Med 1994; 87: 671-8 [ Links ]
19. Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener's granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum 2001; 44: 912-20. [ Links ]
20. Di Benedetto N, López Mujica MJ, Fernández ME, Tourón M, Muñoz SA, Allievi A. Características generales de 29 pacientes con vasculitis de pequeños vasos. Medicina (B Aires) 2010; 70: 127-32. [ Links ]
21. The Wegener´s Granulomatosis Etanercept Trial Research Group. Limited versus Severe Wegener Granulomatosis. Baseline data on patients in the Wegener´s Granulomatosis Etanercept Trial. Arthritis Rheum 2003; 48: 2299-309. [ Links ]
22. Sergent JS. Vasculitis Associated with Antineutrophil Cytoplasmic Antibody. En: Kelley´s textbook of Rheumatology, Sixth Edition; Philadelphia:W. B. Saunders Company. 2001; vol. 2: 1165- 84. [ Links ]
23. Cordier JF, Cottin V. Alveolar hemorrage in vasculitis: primary and secondary. Semin Respir Crit Care Med 2011; 32: 310-21 [ Links ]
24. Eisenberger U, Fakhouri F, Vanhille P, et al. ANCA-negative pauci-immune renal vasculitis: histology and outcome. Nephrol Dial Transplant 2005; 20: 1392-9. [ Links ]
25. MacFarlane DG, Bourne JT, Dieppe PA, Easty DL. Indolent Wegener S granulomatosis. Ann Rheum Dis 1983; 42: 398-407. [ Links ]
26. Holle JU, Gross WL, Holl-Ulrich K, et al. Prospective long-term follow-up of patients with localized Wegener's granulomatosis: does it occur as persistent disease stage? Ann Rheum Dis 2010; 69. 1934-9. [ Links ]
27. Taylor SR, Salama AD, Joshi L, Pusey CD, Lightman SL. Rituximab is effective in the treatment of refractory ophthalmic Wegener´s Granulomatosis. Arthritis Rheum 2009; 60: 1540-7. [ Links ]
Recibido: 7-VI-2012
Aceptado: 6-XI-2012

