










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680
Medicina (B. Aires) vol.73 no.4 Ciudad Autónoma de Buenos Aires July/Aug. 2013
ARTÍCULO ESPECIAL
El uso de alteraciones genéticas en la estratificación por riesgo del mieloma múltiple
Esteban Braggio1, Flavio Albarracín Garramuño2
1Department of Hematology and Oncology, Mayo Clinic, Scottsdale, Arizona, USA,
2Medicina Interna, Hematología, Hospital Universitario, Mendoza, Argentina
Dirección postal: Dr. Flavio Albarracín Garramuño, Olascoaga 2525, 5500 Mendoza, Argentina
e-mail: fhalbarracin@gmail.com
Resumen
Los estudios genéticos han alcanzado un papel central en el estudio del mieloma múltiple (MM), al convertirse en un componente crítico en la estratificación basada en el riesgo de la enfermedad. Se han hecho grandes esfuerzos para identificar cambios genéticos que puedan predecir el resultado clínico e incluirlos en la práctica clínica diaria. La hibridización in situ fluorescente (FISH) es todavía la técnica genética más utilizada en la práctica clínica, mayormente debido a su sencilla implementación y su simplicidad para el análisis de datos. El advenimiento de la genómica (hibridización genómica comparativa, secuenciación exónica o genómica completa) y del transcriptoma de alta resolución (perfiles de expresión de genes - GEP y secuenciación de ARNm) proveen un análisis exhaustivo de los ya definidos factores pronósticos genéticos y son herramientas útiles para la identificación de potenciales nuevos marcadores pronósticos de enfermedad en el clon tumoral de MM. Más aún, GEP ha sido exitosamente implementado en MM como una herramienta de estratificación de riesgo, siendo la de mayor poder de discriminación de resultados. De todas maneras, algunos aspectos técnicos y logísticos complejos (necesidad de una elevada purificación del clon tumoral, costo de los ensayos y complejidad en los análisis de los datos) deben ser considerados antes de la incorporación definitiva de estas tecnologías de alto rendimiento dentro de los ensayos clínicos de rutina. Hasta entonces, FISH continúa siendo la herramienta estándar para la detección de anormalidades genéticas y de valoración pronóstico de enfermedad.
Palabras clave: Herramientas genéticas; Estratificación de riesgo; Mieloma múltiple
Abstract
Genetic tools for risk-stratification in multiple myeloma. Genetic studies have a central role in the study of multiple myeloma (MM), as they become a critical component in the risk-based stratification of the disease. Significant efforts have been made to identify genetic changes and signatures that can predict clinical outcome and include them in the routine clinical care. Fluorescence in situ hybridization (FISH) still remains the most used genetic technique in clinical practice, mostly due to its very straightforward implementation and the simplicity of data analysis. The advent of high-resolution genomics (i.e. array CGH, exome and whole genome sequencing) and transcriptomics tests (i.e. gene expression profiling - GEP, and mRNA sequencing) provide a comprehensive analysis of the already defined genetic prognostic factors and are helpful tools for the identification of potential novel disease markers on the MM tumor clone. Indeed, GEP has been successfully implemented in MM as a risk-stratification tool, holding the greatest power in outcome discrimination. Nevertheless, some technical and logistic intricacies (need of a highly purified tumor clone, cost of the assay and complexity of data analysis) need to be considered before the definitive incorporation of high-throughput technologies in routine clinical tests. Until then, FISH remains the standard tool for genomic abnormality detection and disease prognostication.
Key words: Genetic tools; Risk-stratification; Multiple myeloma
Es ampliamente aceptado que el mieloma múltiple (MM) es una enfermedad heterogénea con subtipos principales definidos por diversas alteraciones genéticas y epigenéticas en las células plasmáticas clonales1, 2. Estas aberraciones genéticas se han utilizado como base para clasificar la enfermedad, establecer categorías de pronóstico, y en cierto grado, servir como marcadores predictivos de respuesta1, 3.
Aunque los test genéticos no tienen valor diagnóstico, tienen alto valor pronóstico y son fundamentales en la estratificación por riesgo del mieloma. FISH (hibridización in situ fluorescente) continuará siendo la técnica estándar para la detección de alteraciones y la consiguiente estratificación del mieloma en grupos de riesgo, hasta el momento en que los análisis genómicos de alta resolución sean de fácil interpretación y bajo costo. El grupo de pacientes considerado de alto riesgo es el más desafiante a nivel clínico debido a la ausencia de una terapia eficaz que pueda ser utilizada. La identificación de nuevos marcadores genéticos que sean predictivos de respuesta es un objetivo central de la investigación en mieloma, ya que de esa forma se puede cambiar el paradigma del tratamiento hacia una terapia más individualizada asociada con el perfil genético del paciente.
Varias clasificaciones genéticas se utilizan en la estratificación del mieloma, generando hasta 8 subgrupos de enfermedades con distintos niveles de agresividad y pronóstico diferencial1, 4-7. Si bien la mayoría de estos perfiles genéticos son principalmente usados con fines investigativos, algunos se han incorporado con éxito al laboratorio clínico8. Por otro lado, la subdivisión de la enfermedad en múltiples subgrupos pequeños conlleva al riesgo de crear análisis fragmentados que carecen de suficiente poder estadístico. En respuesta a este desafío y en un intento de simplificar la estratificación a la hora de la toma de decisiones clínicas, ha sido desarrollada por la Clínica Mayo una clasificación genética simplificada que segrega a los pacientes con pronóstico estándar (75% de los pacientes) y alto (25%) (www.msmart.org; Fig. 1)9, 10. Si bien no existe en la actualidad un tratamiento específico para el MM de alto riesgo, se han generado tres corolarios importantes: i) ensayos clínicos dirigidos a MM de alto riesgo están siendo desarrollados; ii) la estratificación en categorías de riesgo ha permitido tener un tratamiento individualizado en los pacientes de riesgo estándar, transformando en estos casos al MM en una enfermedad crónica; y iii) la identificación de características de alto riesgo es fundamental para la orientación adecuada de los pacientes con un modelo de toma de decisiones compartidas.
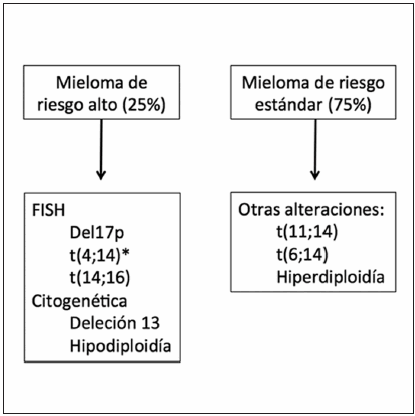
Figura 1 - Estratificación de mieloma por riesgo basado en la presencia de alteraciones genéticas con valor pronóstico (adaptado de mSMART, www.msmart.org).
*Algunos autores la consideran de pronóstico intermedio por su buena respuesta a bortezomib.
El uso de la estratificación basada en el perfil genético se ha utilizado tanto en pacientes tratados con quimioterapia convencional4, como con quimioterapia a altas dosis seguida por trasplante autólogo de células progenitoras hematopoyéticas1, 7. Se están identificando perfiles genéticos predictivos de respuesta a nuevas terapias, basadas en inhibidores del proteasoma y drogas inmunomodulatorias (IMiDs)11, 13, con el fin de cambiar el paradigma del tratamiento desde las terapias actuales a las terapias paciente-específicas, dependiendo de su perfil genético. Un ejemplo es la identificación del subgrupo de pacientes con bajo nivel de expresión génica del gen cereblon (CRBN). Recientemente fue descubierto que la talidomida inicia sus efectos teratogénicos al unirse a CRBN e inhibir la actividad ubiquitina ligasa al complejo E3, constituido por CRBN, Cul4A y damage DNA binding protein 1 (DDB1)14. Posteriormente se demostró que CRBN es indispensable para la función de los IMiDs15. De esta forma, los casos con niveles bajos de CRBN no se beneficiarían con terapias basadas en IMiDs, y otras terapias deberían ser consideradas.
Antecedentes de estudios genéticos en MM
En los últimos 30 años los estudios moleculares comenzaron a revelar alteraciones cromosómicas en MM que ayudan a identificar la enfermedad más agresiva16-18. Esta clasificación biológica es impulsada principalmente por alteraciones genéticas que se pueden observar desde las fases pre-malignas de la enfermedad19, 20. A gran escala, los mielomas se dividen en hiperdiploides y no hiperdiploides. Este último grupo a su vez se subdivide en hipodiploides (< 45 cromosomas), pseudodiploides (45 a 47 cromosomas) y casi tetraploides (> 73 cromosomas). Cada uno de estos grupos (MM hiperdiploide y no hiperdiploide) comprende aproximadamente la mitad de los casos con muy bajo solapamiento entre categorías2. Los casos hiperdiploides están caracterizados por la presencia de múltiples trisomías, especialmente de los cromosomas impares 3, 5, 7, 9, 11, 15, 19 y 21. Por otro lado, los no-hiperdiploides están caracterizados principalmente por la presencia de translocaciones cromosómicas afectando el locus IgH en la banda 14q324, 21, 22. La mayoría de estas translocaciones conducen a la activación de proto-oncogenes ubicados en las bandas 11p13 (cyclin D1 [CCND1; encontrado en 15% de los pacientes), 4p16 (Wolf-Hirschhorn sindrome candidate 1 [MMSET] y fibroblast growth factor receptor 3 [FGFR3]; 15%), 16q23 (v-maf musculoaponeurotic fibrosarcoma oncogene homolog [MAF]; 5%), 6p21 (cyclin D3 [CCND3]; < 5%) y 20q12 (v-maf musculoaponeurotic fibrosarcoma oncogene homolog B [MAFB]; < 5%)23-25. Todas estas translocaciones tienen valor pronóstico, como se discute en la próxima sección.
Aunque existen muchos sistemas que pueden estimar el pronóstico del MM, la mayoría de los enfoques se centran en la identificación de marcadores genéticos que pueden discriminar entre grupos clínicos. La genética ha sido utilizada por varios estudios a gran escala para predecir el resultado en los pacientes tratados con terapia convencional4, 26 y dosis alta de quimioterapia seguida de trasplante autólogo de células progenitoras hematopoyéticas (TACPH)1, 7. La mayoría de estos estudios utilizaron agentes alquilantes. En la actualidad se están realizando varios estudios para comprender mejor las implicaciones pronósticas de marcadores genéticos en cohortes de pacientes tratados con inhibidores del proteasoma (bortezomib y carfilzomib) e IMiDs (lenalidomida y pomalidomida).
Aunque todavía no existen datos concluyentes sobre el valor pronóstico de los marcadores genéticos con estos nuevos esquemas terapéuticos, los datos preliminares sugieren que tanto el tratamiento basado en bortezomib12, 27, 28 como en talidomida y lenalidomida11, 29, 30, podría beneficiar a los que poseen marcadores genéticos de alto riesgo. A pesar del entusiasmo inicial, estos estudios han sido realizados con grupos reducidos de pacientes, y la mayoría en el marco de MM reincidente y refractario12, 29. También es importante señalar que el valor pronóstico de los marcadores genéticos necesita ser validado de acuerdo con la etapa específica de la enfermedad; factores pronósticos validados en el tratamiento inicial de la enfermedad pueden no tener efectos similares en pacientes previamente tratados.
El campo de estudio se desarrolló inicialmente por el uso de FISH1, 4, 21, 31, y más recientemente por medio de plataformas de alta resolución para el estudio del transcriptoma [perfiles de expresión génica (GEP)])6, 7, 32 y genoma [hibridación genómica comparada (aCGH) y secuenciación de segunda generación]5, 33, 34. Es fundamental que los análisis genómicos se hagan exclusivamente en células tumorales para asegurar resultados precisos y fiables. La identificación de células tumorales puede ser realizada por medio de dos métodos. El primero consta de la identificación de las células plasmáticas clonales por medio de la marcación fluorescente de moléculas de inmunoglobulina de cadena liviana localizadas en el citoplasma de las células plasmáticas (cIg)35. Otra opción es enriquecer las células plasmáticas por medio de perlas magnéticas anti-CD138. La cIg es exclusivamente usada en combinación con FISH (cIg-FISH), mientras que la purificación de células plasmáticas puede ser usada tanto para FISH como para estudios genómicos.
Alteraciones genéticas con valor pronóstico
Translocación t(4;14)(p16;q32)
La translocación t(4;14) es observada en 15% de los pacientes y resulta en la sobre-expresión de los genes FGFR3 y MMSET. La translocación está asociada con una enfermedad más agresiva y supervivencia reducida, tanto al diagnóstico como en pacientes tratados con quimioterapia basada en alquilantes o con TACPH1, 3, 4, 7. Si bien los datos iniciales mostraron que los pacientes con esta translocación responden mejor al tratamiento con bortezomib que a terapias convencionales12, estudios posteriores en grandes cohortes tratadas con bortezomib indican que la translocación todavía muestra valor pronóstico, en series de pacientes analizadas por los grupos multicéntricos francés (grupo IFM)30 y español3, 10, 36. De hecho, la serie francesa muestra que si bien el bortezomib ha mejorado el resultado en casos con t(4;14) en comparación con terapias previas, el marcador genético está todavía asociado con un pronóstico negativo. Por su lado, el estudio español ha mostrado resultados similares y confirma que la t(4;14) todavía augura un pronóstico desfavorable en MM.
Translocación t(14;16)(q32;q23) y otras alteraciones de MAF
La t(14;16) es identificada en 5% de los casos y resulta en la sobre-expresión del proto-oncogén MAF. Pacientes con la t(14;16) presentan una enfermedad agresiva y mal pronóstico, independientemente del esquema terapéutico utilizado4, 7. Un resultado similar se asocia con la t(14;20) que afecta al gen MAFB37. La detección de estas translocaciones no ha sido universalmente establecida en el laboratorio clínico dada su muy baja prevalencia. Un estudio reciente del IFM ha cuestionado la importancia pronóstica de la t(14;16), dado que un gran número de casos mostraron un efecto neutral sobre la misma13. Sin embargo, también se identificó que el MM con t(14;16) tenía una alta propensión a mostrar muchas células circulantes, un sello distintivo del MM agresivo.
Translocación t(11;14)(q13;q32)
La t(11;14) está asociada con baja proliferación celular, bajo nivel de expresión de CD20 en superficie, bajos niveles séricos de proteínas monoclonales y pronóstico favorable4. Los casos con t(11;14) son caracterizados por células con una morfología linfoplasmacítica y mayor compromiso óseo38, 39. Esta translocación se encuentra en una alta proporción con la variante de IgM (38%), así como la amiloidosis de cadena de inmunoglobulina ligera. Adicionalmente, mutaciones de K-RAS son identificadas en el 50% de los casos con t(11;14) comparado con 10% en casos con otras translocaciones del locus IgH40.
Deleción 17p13
La deleción 17p13, presente en el 5-10% de los casos, sigue siendo el factor genético de pronóstico desfavorable más importante en mieloma1, 4, 7, 26. El valor pronóstico se observa tanto en los esquemas quimioterápicos convencionales como con TACPH, bortezomib o lenolidomida, lo que sugiere que ninguno de los tratamientos tiene un impacto significativo en este subgrupo11, 30. Los datos de las series clínicas más recientes indican que mientras las nuevas terapias han mejorado las perspectivas, estos beneficios se distribuyen de forma desigual, siendo mínima con la deleción 17p, moderado para t(4;14) y mayor para la enfermedad de riesgo estándar.
Aunque el gen que causa el efecto negativo no ha sido completamente confirmado, toda la evidencia indica que TP53 es el candidato principal. Los casos con deleción 17p13, a menudo tienen enfermedad extramedular, hipercalcemia, afectación del sistema nervioso central y otras características de enfermedad más agresiva3, 4, 41.
Amplificación de 1q
Múltiples estudios han demostrado que alteraciones del cromosoma 1q se asocian con un peor pronóstico en MM42-44. El principal gen candidato localizado en 1q es CDC28 protein kinase regulatory subunit 1B (CKS1B), el cual promueve la degradación de p27, un inhibidor de la progresión del ciclo celular. Existen dos perfiles de expresión genética usados en la estratificación por riesgo de pacientes con MM, de los cuales un 30% de los genes incluidos en el perfil se encuentra en el cromosoma 117. Aunque la asociación entre amplificación 1q y mal pronóstico ha sido demostrada en mieloma44, su análisis todavía no se ha implementado en la práctica clínica estándar.
La deleción de 1p también se observa en una proporción considerable de casos con MM. Sin embargo, todavía no ha sido identificado un gen candidato en esa región.
Deleción 13
La deleción del cromosoma 13 es una de las alteraciones cromosómicas más comunes en MM, identificada en casi el 50% de los pacientes recién diagnosticados21, 45-48. Varios estudios han demostrado una prevalencia similar en MGUS, lo que indica que se trata de un evento inicial en la patogénesis de la enfermedad19, 20, 49. La deleción fue originalmente identificada como un indicador de pronóstico negativo17. Sin embargo, cuando la alteración es identificada mediante FISH, el impacto no es lo suficientemente fuerte como para considerar la aberración indicativa de alto riesgo, sino más bien como de riesgo intermedio4, por lo que su valor clínico se considera sólo cuando la deleción está presente en el estudio citogenético convencional8. La explicación más probable de la diferencia entre técnicas de la detección de la deleción 13q, es que la identificación de la alteración en las metafases es un indicador de una enfermedad más proliferativa y con mayor carga tumoral. Por lo tanto, el valor pronóstico se debe a su función como marcador de proliferación celular más que a un efecto directo ejercido por dicha alteración. Adicionalmente, la deleción 13 está comúnmente asociada con marcadores genéticos de alto riesgo, tales como t(4;14) y deleción 17p2.
Estudios genómicos de alta resolución
Los estudios genómicos de alta resolución son ideales en el estudio del mieloma, ya que no solo proporcionan un análisis completo de los factores pronósticos ya definidos, sino que también son herramientas útiles para la identificación de nuevos marcadores de la patología. Sin embargo, algunos puntos deben ser considerados antes de su incorporación en los ensayos clínicos de rutina. Como ya se mencionó, el análisis se debe realizar exclusivamente en células tumorales, ya sea identificándolas en combinación con inmunofluorescencia de la cadena Ig ligera citoplasmática35, o bien siendo purificadas usando anticuerpos anti-CD138+. Adicionalmente, otros puntos a tener en cuenta son el alto costo de los ensayos y la mayor complejidad de los análisis asociados.
Perfil de expresión génica (GEP)
El uso del GEP como una herramienta de estratificación de riesgo se ha aplicado con éxito en mieloma6, 7, 50. A partir de dos grandes series de pacientes tratados homogéneamente (Total Therapy 2 y Total Therapy 3), investigadores de la universidad de Arkansas identificaron dos sets de genes que sirven para estratificar a pacientes en grupos de bajo y alto riesgo7. Algunos de los genes más representativos de esa lista son: CKS1B, thymopoietin (TMPO), latent transforming growth factor beta binding protein 1 (LTBP1), WEE1 y baculoviral IAP repeat containing 5 (BIRC5), entre otros. Estos se correlacionan con otros marcadores de enfermedad agresiva, como la proliferación celular. Estos sets de genes no han sido todavía validados en pacientes tratados con inhibidores del proteasoma e IMiDs, aunque probablemente tengan el mismo valor que para las otras cohortes. El GEP también es muy exacto en la detección de translocaciones de IgH, debido a la identificación de muy altos niveles de expresión de los proto-oncogenes involucrados en las distintas translocaciones50. Infortunadamente, el GEP no puede predecir la presencia de la deleción 17p13, uno de los factores pronósticos más adversos en mieloma.
Hibridización genómica comparativa en array (array-CGH).
Esta tecnología tiene la ventaja de trabajar con el ADN, evitando de este modo las limitaciones relacionadas con la estabilidad del ARN. Estudios recientes han utilizado plataformas de array-CGH y SNP (polimorfismo de nucleótido simple) en un intento de incorporar herramientas de valor pronóstico para MM5, 33. Uno de estos estudios propuso una clasificación en 3 subgrupos pronósticos basada en la combinación del nivel de ß2 microglobulina y nuevas alteraciones genómicas con valor pronóstico, como ganancia de 5q y deleción de 12p33. Sin embargo, se necesitan más estudios antes de su aplicación en la práctica clínica.
Una limitación importante de estas plataformas es que no están diseñadas para identificar translocaciones, por lo tanto su uso no puede reemplazar a FISH y GEP, sino más bien complementarlos.
Secuenciación completa del genoma
La secuenciación completa del genoma está revolucionando el estudio del cáncer. Los primeros 38 mielomas han sido secuenciados y se ha confirmado la alta frecuencia de mutaciones afectando a la vía NF-kB, así como también genes responsables de la traducción proteica34. Este enfoque es muy caro y el análisis es extremadamente complicado como para ser considerado como una prueba de rutina en el análisis del mieloma; sin embargo, su implementación en la clínica podrá acontecer en un futuro no tan lejano.
Aspectos prácticos y pruebas genéticas recomendadas
La estratificación en grupos de riesgo basada en alteraciones genéticas puede tener profundas consecuencias en las decisiones clínicas y terapéuticas10. Inicialmente, las pruebas se realizaron usando citogenética convencional; sin embargo, el escaso éxito en la obtención de células tumorales en división llevó a su casi completa erradicación del laboratorio clínico. La herramienta genética ideal de pronóstico debe: i) ser confiable en la predicción de resultados; ii) poder ser fácilmente implementada en el laboratorio clínico; y iii) estar ampliamente disponible y fácil de interpretar. El FISH cumple la mayoría de estos requisitos, siendo una técnica muy sencilla de implementar y analizar. Es por todo esto, que todavía hoy el FISH sigue siendo la herramienta de diagnóstico primario. Por otro lado, la técnica de FISH tiene sus limitaciones, especialmente por el hecho de analizar solo una o pocas regiones cromosómicas, careciendo de una perspectiva integral de la complejidad genómica del tumor. Su asignación categórica de los pacientes en subgrupos es, por lo tanto, intrínsecamente limitada y en general sólo puede proporcionar una orientación general.
El GEP, por otro lado, genera mayor cantidad de datos y su uso se ha empezado a masificar en los laboratorios clínicos y es probable que reemplace al FISH como la prueba estándar en la clínica. Para ciertas alteraciones, como la deleción 17p13, la transición de FISH a GEP no es tan sencilla y aún deben desarrollarse más opciones antes de la eliminación de la prueba en curso (Tabla 1). Aunque, sin duda, esto cambie con el tiempo, puede ser utilizado como una guía general sobre qué estudio realizar y en qué punto de la enfermedad.
TABLA 1.- Sumario del tipo de test útil para el estudio de alteraciones con valor pronóstico
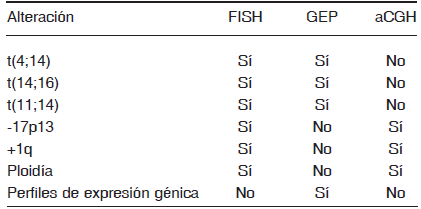
A nuestro entender, la mejor estrategia actualmente disponible para implementar en el laboratorio clínico es el uso de GEP para la detección de translocaciones e índices genéticos utilizados en estratificación por riesgo, acompañado con el estudio de 17p mediante la técnica de FISH. Sin embargo, hasta el momento en que el GEP y otras técnicas de alta resolución, como la secuenciación del genoma o transcriptoma, se conviertan en herramientas clínicas estándar, FISH continuará siendo la principal herramienta para establecer criterios pronósticos moleculares en MM.
Agradecimientos: Los autores agradecen a Henry Predolin Foundation, The Marriott Specialized Workforce Development Awards in Individualized Medicine, y a George Haub Family Career Development Award Fund in Cancer Research, por el soporte recibido.
Conflictos de interés: Los autores no tienen conflictos de interés para declarar.
1. Avet-Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood 2007; 109: 3489-95. [ Links ]
2. Fonseca R, Barlogie B, Bataille R, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res 2004; 64: 1546-58. [ Links ]
3. Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia 2009; 23: 2210-21. [ Links ]
4. Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003; 101: 4569-75. [ Links ]
5. Carrasco DR, Tonon G, Huang Y, et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell 2006; 9: 313-25. [ Links ]
6. Chng WJ, Braggio E, Mulligan G, et al. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood 2008; 111: 1603-9. [ Links ]
7. Shaughnessy JD, Jr., Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007; 109: 2276-84. [ Links ]
8. Fonseca R, San Miguel J. Prognostic factors and staging in multiple myeloma. Hematol Oncol Clin North Am 2007; 21: 1115-40, ix. [ Links ]
9. Dispenzieri A, Rajkumar SV, Gertz MA, et al. Treatment of newly diagnosed multiple myeloma based on Mayo Stratification of Myeloma and Risk-adapted Therapy (mSMART): consensus statement. Mayo Clin Proc 2007; 82: 323-41. [ Links ]
10. Kumar SK, Mikhael JR, Buadi FK, et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus guidelines. Mayo Clin Proc 2009; 84: 1095-110. [ Links ]
11. Reece D, Song KW, Fu T, Roland B, et al. Influence of cytogenetics in patients with relapsed or refractory multiple myeloma treated with lenalidomide plus dexamethasone: adverse effect of deletion 17p13. Blood 2009; 114: 522-5. [ Links ]
12. San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008; 359: 906-17. [ Links ]
13. Avet-Loiseau H, Malard F, Campion L, et al. Translocation t(14;16) and multiple myeloma: is it really an independent prognostic factor? Blood 2011; 117: 2009-11. [ Links ]
14. Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science 2010; 327: 1345-50. [ Links ]
15. Zhu YX, Braggio E, Shi CX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011; 118: 4771-9. [ Links ]
16. Dewald GW, Kyle RA, Hicks GA, Greipp PR. The clinical significance of cytogenetic studies in 100 patients with multiple myeloma, plasma cell leukemia, or amyloidosis. Blood 1985; 66: 380-90. [ Links ]
17. Tricot G, Barlogie B, Jagannath S, et al. Poor prognosis in multiple myeloma is associated only with partial or complete deletions of chromosome 13 or abnormalities involving 11q and not with other karyotype abnormalities. Blood 1995; 86: 4250-6. [ Links ]
18. Tricot G, Sawyer JR, Jagannath S, et al. Unique role of cytogenetics in the prognosis of patients with myeloma receiving high-dose therapy and autotransplants. J Clin Oncol 1997; 15: 2659-66. [ Links ]
19. Avet-Loiseau H, Facon T, Daviet A, et al. 14q32 translocations and monosomy 13 observed in monoclonal gammopathy of undetermined significance delineate a multistep process for the oncogenesis of multiple myeloma. Intergroupe Francophone du Myelome. Cancer Res 1999; 59: 4546-50. [ Links ]
20. Fonseca R, Bailey RJ, Ahmann GJ, et al. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood 2002; 100: 1417-24. [ Links ]
21. Debes-Marun CS, Dewald GW, Bryant S, et al. Chromosome abnormalities clustering and its implications for pathogenesis and prognosis in myeloma. Leukemia 2003; 17: 427-36. [ Links ]
22. Smadja NV, Fruchart C, Isnard F, et al. Chromosomal analysis in multiple myeloma: cytogenetic evidence of two different diseases. Leukemia 1998; 12: 960-9. [ Links ]
23. Chesi M, Bergsagel PL, Shonukan OO, et al. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood 1998; 91: 4457-63. [ Links ]
24. Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 1998; 92: 3025-34. [ Links ]
25. Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene 2001; 20: 5611-22. [ Links ]
26. Drach J, Ackermann J, Fritz E, et al. Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood 1998; 92: 802-9. [ Links ]
27. Jagannath S, Richardson PG, Sonneveld P, et al. Bortezomib appears to overcome the poor prognosis conferred by chromosome 13 deletion in phase 2 and 3 trials. Leukemia 2007; 21: 151-7. [ Links ]
28. Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352: 2487-98. [ Links ]
29. Kapoor P, Kumar S, Fonseca R, et al. Impact of risk stratification on outcome among patients with multiple myeloma receiving initial therapy with lenalidomide and dexamethasone. Blood 2009; 114: 518-21. [ Links ]
30. Avet-Loiseau H, Soulier J, Fermand JP, et al. Impact of high-risk cytogenetics and prior therapy on outcomes in patients with advanced relapsed or refractory multiple myeloma treated with lenalidomide plus dexamethasone. Leukemia 2010; 24: 623-8. [ Links ]
31. Drach J, Schuster J, Nowotny H, et al. Multiple myeloma: high incidence of chromosomal aneuploidy as detected by interphase fluorescence in situ hybridization. Cancer Res 1995; 55: 3854-9. [ Links ]
32. Decaux O, Lode L, Magrangeas F, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myelome. J Clin Oncol 2008; 26: 4798-805. [ Links ]
33. Avet-Loiseau H, Li C, Magrangeas F, Gouraud W, et al. Prognostic significance of copy-number alterations in multiple myeloma. J Clin Oncol 2009; 27: 4585-90. [ Links ]
34. Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471: 467-72. [ Links ]
35. Ahmann GJ, Jalal SM, Juneau AL, et al. A novel three-color, clone-specific fluorescence in situ hybridization procedure for monoclonal gammopathies. Cancer Genet Cytogenet 1998; 101: 7-11. [ Links ]
36. Mateos M, Gutierrez N, Paiva B, et al. Clinical Outcome According to Both Cytogenetic Abnormalities (CA) Detected by Fluorescence In Situ Hibridization (FISH) and Hyperdiploidy Assessed by Flow Cytometry (FCM) In Elderly Newly Diagnosed Myeloma Patients Treated with A Bortezomib-Based Combination. Blood 2010: Abstract 309. [ Links ]
37. Ross FM, Chiecchio L, Dagrada G, et al. The t(14;20) is a poor prognostic factor in myeloma but is associated with long-term stable disease in monoclonal gammopathies of undetermined significance. Haematologica 95: 1221-5. [ Links ]
38. Fonseca R, Blood EA, Oken MM, et al. Myeloma and the t(11;14)(q13;q32); evidence for a biologically defined unique subset of patients. Blood 2002; 99: 3735-41. [ Links ]
39. Garand R, Avet-Loiseau H, Accard F, Moreau P, Harousseau JL, Bataille R. t(11;14) and t(4;14) translocations correlated with mature lymphoplasmacytoid and immature morphology, respectively, in multiple myeloma. Leukemia 2003; 17: 2032-5. [ Links ]
40. Rasmussen T, Kuehl M, Lodahl M, Johnsen HE, Dahl IM. Possible roles for activating RAS mutations in the MGUS to MM transition and in the intramedullary to extramedullary transition in some plasma cell tumors. Blood 2005; 105: 317-23. [ Links ]
41. Chng WJ, Price-Troska T, Gonzalez-Paz N, et al. Clinical significance of TP53 mutation in myeloma. Leukemia 2007; 21: 582-4. [ Links ]
42. Chang H, Qi X, Trieu Y, et al. Multiple myeloma patients with CKS1B gene amplification have a shorter progression-free survival post-autologous stem cell transplantation. Br J Haematol 2006; 135: 486-91. [ Links ]
43. Fonseca R, Van Wier SA, Chng WJ, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia 2006; 20: 2034-40. [ Links ]
44. Zhan F, Colla S, Wu X, et al. CKS1B, overexpressed in aggressive disease, regulates multiple myeloma growth and survival through SKP2- and p27Kip1-dependent and -independent mechanisms. Blood 2007; 109: 4995-5001. [ Links ]
45. Avet-Loiseau H, Facon T, Grosbois B, et al. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood 2002; 99: 2185-91. [ Links ]
46. Avet-Louseau H, Daviet A, Sauner S, Bataille R. Chromosome 13 abnormalities in multiple myeloma are mostly monosomy 13. Br J Haematol 2000; 111: 1116-7. [ Links ]
47. Desikan R, Barlogie B, Sawyer J, Ayers D, Tricot G, Badros A, et al. Results of high-dose therapy for 1000 patients with multiple myeloma: durable complete remissions and superior survival in the absence of chromosome 13 abnormalities. Blood 2000; 95: 4008-10. [ Links ]
48. Facon T, Avet-Loiseau H, Guillerm G, et al. Chromosome 13 abnormalities identified by FISH analysis and serum beta2-microglobulin produce a powerful myeloma staging system for patients receiving high-dose therapy. Blood 2001; 97: 1566-71. [ Links ]
49. Drach J, Angerler J, Schuster J, et al. Interphase fluorescence in situ hybridization identifies chromosomal abnormalities in plasma cells from patients with monoclonal gammopathy of undetermined significance. Blood 1995; 86: 3915-21. [ Links ]
50. Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J, Jr. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood 2005; 106: 296-303. [ Links ]
Recibido: 19-XI-2012
Aceptado: 20-III-2013

