










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680
Medicina (B. Aires) vol.73 supl.1 Ciudad Autónoma de Buenos Aires Sept. 2013
ACTUALIZACIÓN EN NEUROLOGÍA INFANTIL IV
Orientación diagnóstica de las enfermedades neurometabólicas basada en la clínica, estudios metabólicos y neuroimagenológicos
Jaume Campistol
Servicio de Neuropediatría, Hospital Universitario Sant Joan de Deu, Barcelona, España
Dirección postal: Dr. Jaume Campistol, Servicio de Neuropediatría, Passeig Sant Joan de Deu s/n, Hospital Universitari Sant Joan de Deu, Esplugues de Llobregat 08950, Barcelona, España
e-mail: campistol@hsjdbcn.es
Resumen
Las enfermedades neurometabólicas constituyen un grupo de patologías raras en notable expansión, de difícil diagnóstico y manejo. Este artículo pretende desarrollar una orientación práctica frente a la sospecha de una enfermedad metabólica, obtener datos del examen clínico, de los estudios bioquímicos y de las técnicas de neuroimagen orientadoras. Es muy importante para el neuropediatra sospecharlas siempre en función de los signos y síntomas neurológicos como el retardo del desarrollo, déficit cognitivo, epilepsia refractaria, distonía, crisis metabólicas o la presencia de signos extraneurológicos inexplicados. También es importante el diagnóstico de cara a las nuevas opciones terapéuticas, consejo genético y diagnóstico prenatal.
Palabra clave: Enfermedades neurometabólicas; Signos neurológicos; Bioquímica; Neuroimagen; Tratamiento
Abstract
Guidelines for detection of inborn errors of metabolism based on clinical exam, analytical studies and neuroimaging techniques. Neurometabolic disorders constitute an expanding and complex field in which it is difficult to diagnose and to acquire a specific education and training. This article tries to develop a practical orientation in the suspicion, clinical exam, biochemical studies and neuroimaging techniques for the detection of inborn errors of metabolism. It is very important for the neuropediatrician to suspect metabolic diseases depending on some of the most frequent unexplained neurological disturbances and symptoms as psychomotor delay, mental retardation, refractory epilepsy, dystonia, metabolic crisis or other extraneurological signs. It is important the diagnosis related to the new emergent therapeutic options, genetic counseling and prenatal diagnosis.
Key words: Neurometabolic diseases; Neurological signs; Biochemical; Neuroimaging; Treatment
Los errores congénitos del metabolismo (ECM) están causados por mutaciones del DNA que generan proteínas anómalas en las que la estructura y por tanto la función está alterada. Cuando estas proteínas mutadas interfieren en los procesos de síntesis, catabolismo o transporte de pequeñas moléculas (aminoácidos, ácidos grasos, hidratos de carbono) o en las reacciones de producción de energía, los denominamos ECM1.
A lo largo de las últimas tres décadas, los ECM han adquirido un papel preponderante dentro de la patología pediátrica. La mejor formación de los profesionales y los avances tecnológicos tanto en el ámbito bioquímico como en el genético, han permitido un mejor conocimiento de los ECM. Contribuye además la descripción de un número creciente de enfermedades metabólicas congénitas, el poder caracterizar muchas de las proteínas anómalas que constituyen el origen bioquímico del defecto y el conocer las mutaciones causantes de un número considerable de ECM. Se han descrito nuevas categorías de enfermedades metabólicas asociadas a las diferentes organelas celulares, como las enfermedades del metabolismo energético mitocondrial, enfermedades peroxisomales, defectos localizados en el retículo endotelial y aparato de Golgi, defectos de la creatina cerebral, defectos de la glicosilación de proteínas, defectos de los neurotransmisores, metales, fosfolípidos, etc.1.
Aunque considerados individualmente los ECM son enfermedades raras, en su conjunto constituyen más de 5% de las admisiones en un hospital pediátrico, y una frecuencia en la población general de cerca de 1/2 000 nacidos vivos2, 3. Los ECM son una causa frecuente de mortalidad y morbilidad en la infancia1, 2. Todo ello ha determinado que muchos hospitales pediátricos dedicaran una atención especial a este ámbito de la patología infantil, no sólo al diagnóstico sino también al tratamiento específico de los pacientes. La colaboración de laboratorios de referencia especializados en el estudio bioquímico/genético ha resultado muy valiosa para el diagnóstico definitivo de muchos pacientes con ECM.
Se han descrito un gran número de ECM1, 2, 4. Algunos de los cuales con un número importante de observaciones (fenilcetonuria) y otros con casos únicos o muy aislados.
La incidencia exacta es difícil de precisar ya que hay escasos datos estadísticos de frecuencia en la población general y solamente se estudian en sujetos con sintomatología. Con la aparición del cribado metabólico neonatal expandido se ha incrementado el número de diagnósticos y se conoce mejor la incidencia de muchas de estas enfermedades. De la precocidad y precisión en el diagnóstico dependerá el tratamiento efectivo en muchos casos, evitando no solamente la muerte, sino secuelas neurológicas irreversibles.
Fisiopatología
Desde el punto de vista fisiopatológico la mayor parte de los ECM que producen una clínica aguda y que con frecuencia precisan atención en unidades de cuidados intensivos pediátricos corresponden a los ECM intermediario. Se conocen tres bases fisiopatológicas principales:
1) Intoxicación aguda o progresiva secundaria al acúmulo de productos tóxicos próximos o relacionados con el bloqueo metabólico. Dentro de este grupo incluiríamos las aminoacidopatías, acidemias orgánicas, deficiencias del ciclo de la urea e intolerancia a los hidratos de carbono.
2) Deficiencia en la producción de energía y/o utilización de substratos energéticos que se manifestará principalmente en los órganos o tejidos con más requerimientos (SNC, músculo, hígado, riñón) y producen enfermedades multisistémicas y miopatías. Dentro de este grupo se incluyen los defectos de transporte de substratos energéticos en la mitocondria (piruvato, carnitina), defectos de metabolismo del piruvato, del ciclo de Krebs, de la cadena respiratoria, de la beta-oxidación de ácidos grasos y de la gluconeogénesis.
3) Defectos que se manifiestan principalmente como hepatopatías agudas; en este grupo se incluyen las glucogenosis hepáticas I-III, VI y IX, tirosinemia I, galactosemia, intolerancia a la fructosa, deficiencia de fructosa 1-6 difosfatasa, defectos de la beta-oxidación de ácidos grasos, deficiencia de alfa-1 antitripsina y los defectos de la cadena respiratoria.
Orientación diagnóstica
La orientación diagnóstica no siempre es sencilla y las manifestaciones clínicas de un ECM pueden ser de lo más variado posible desde una simple sordera, convulsiones, deterioro neurológico, coma o disfunción hepática aguda. Se basa pues en tres pilares fundamentales (a, b, c) y la confirmación diagnóstica en los otros dos puntos básicos (d, e)1, 5 (Tabla 1).
TABLA 1.- Orientación y confirmación de un ECM

Es muy importante una buena anamnesis (analizando datos positivos de consanguinidad, familiares próximos con un problema similar), un examen clínico detallado y análisis de todos los datos de laboratorio disponibles. La sintomatología clínica es en general bastante inespecífica, no solo de la vía metabólica afectada sino incluso de la existencia de un defecto congénito. La alteración de alguno de los parámetros analíticos asociada a sintomatología clínica y datos anamnésicos positivos orienta fuertemente hacia un ECM. Sin embargo, la normalidad de todos estos datos no excluye en absoluto la posibilidad de un ECM siempre que la clínica así lo haga sospechar. Muchas veces la sospecha diagnóstica es por exclusión de otras enfermedades mucho más prevalentes que los ECM.
Es muy importante la estrecha colaboración entre el clínico y el bioquímico clínico para poder llegar a un diagnóstico, ya que éste no viene dado por un valor único de una magnitud bioquímica o un dato semiológico sino de la interpretación de un conjunto de datos clínicos, analíticos y de la experiencia del equipo. La descompensación metabólica sin apenas motivo puede ser una indicación hacia defecto del metabolismo intermediario. Es muy importante que las muestras, en las formas de presentación aguda se tomen en períodos de descompensación, y muy especialmente antes de comenzar cualquier tratamiento.
Formas de presentación de los ECM
Hemos dividido los ECM según su forma de presentación en agudos y subagudos o crónicos y con manifestaciones clínicas particulares (Tabla 2).
TABLA 2.- Signos clínicos sugestivos de presentación aguda de los ECM
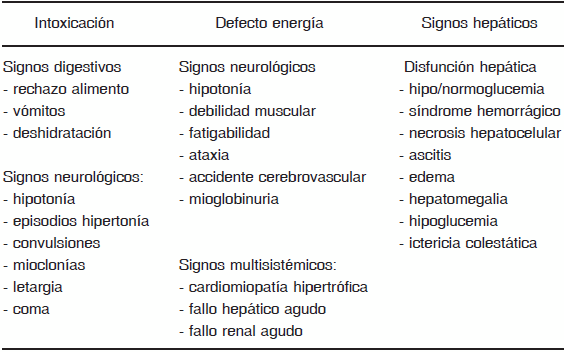
1) Sintomatología clínica de los ECM de presentación aguda
Síntomas clínicos inespecíficos
Alteración brusca del estado general sin motivo aparente
Manifestaciones neurológicas agudas (hipotonía, somnolencia, hiporreactividad, convulsiones)
Trastornos digestivos (rechazo de alimento, vómitos)
Trastornos respiratorios (polipnea, bradipnea, etc.)
Datos evocadores
Antecedentes familiares de muertes neonatales o inexplicables6-8
Consanguinidad
Abortos a repetición
Deterioro progresivo sin causa aparente y que no responde a la terapia específica
Signos clínicos específicos
Deterioro neurológico rápido sin un origen claro (trastorno de conciencia, del tono, movimientos anormales, convulsiones, alteraciones específicas EEG)
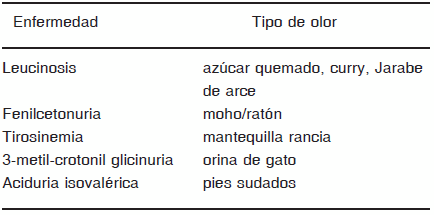
Olor especial (corporal o de la orina) (Tabla 3)
Miopatía - cardiomiopatía (metabolismo energético)
Hepatomegalia
Cataratas, luxación del cristalino
Litiasis renal
Dismorfia cráneo - facial (déficit PDH, acidemia mevalónica, glutárica tipo II, peroxisomales)
TABLA 3.- Olor corporal especial en los ECM
Presentación como disfunción hepática
Hepatomegalia
Ictericia
Vómitos
Letargia
Alteración función hepática
La presencia de una infección no descarta la coexistencia de un ECM, pues con mucha frecuencia la primera es la desencadenante de la sintomatología.
2) ECM que se presentan con manifestaciones clínicas tardías y/o episodios recurrentes de descompensación
Las características de estos ECM son las manifestaciones agudas, la presentación tardía, un curso rápido hacia la mejoría y un nuevo empeoramiento o la muerte de forma inesperada. En general presentan ataques recurrentes de descompensaciones coincidiendo con situaciones de estrés o infecciones. En estos períodos las manifestaciones clínicas y bioquímicas son evidentes; sin embargo, entre los ataques predomina la normalidad clínico-bioquímica. Los defectos congénitos más habituales en este grupo son los trastornos del ciclo de la urea, de la beta-oxidación de los ácidos grasos y cetogénesis, catabolismo de los aminoácidos ramificados, deficiencia de biotinidasa, defectos de glucogenolisis y neoglucogénesis1, 3.
3) ECM con manifestaciones particulares de un solo órgano o sistema
Dermatológicos (dermatitis, alopecia, alteraciones del cabello)
Dismorfológicos (facial, agenesia del cuerpo calloso)
Gastroenterológicos (anorexia, fallo de medro, dolor abdominal, diarrea)
Hematológicos (pancitopenia, neutropenia)
Hepáticos (cirrosis, ictericia)
Nefrológicos (síndrome hemolítico-urémico, nefropatía, síndrome de Fanconi)
Neurológicos (ataxia, distonía, epilepsia, paraparesia espástica, deterioro neurológico)
Oftalmológicos (cataratas, ectopia cristalino)
Óseos (osteoporosis)
Psiquiátricos (autismo, psicosis, histeria, regresión)
Vasculares (tromboembolismo, AVC)
Es muy importante la sospecha diagnóstica , y el clínico debe tener siempre presentes los ECM como una de las causas no habituales de cuadros agudos, que comprometen la vida del paciente y que muchas veces son absolutamente enigmáticos y difíciles de reconocer1, 9, 10.
Estudios bioquímicos para el diagnóstico de errores congénitos del metabolismo
El diagnóstico de los errores congénitos del metabolismo (ECM) se basa del estudio de las alteraciones bioquímicas que muestren los pacientes, previamente seleccionados en base a la sospecha clínica. Las alteraciones bioquímicas comprenden desde leves anomalías de la analítica básica que sugieren la existencia de un ECM y a veces permiten orientar el diagnóstico, hasta el estudio de los marcadores bioquímicos específicos (metabolitos, actividades enzimáticas, caracterización de proteínas) de cada una de estas enfermedades (Tablas 4 y 5). Será necesario disponer de un protocolo de recogida de muestras ante la sospecha de un ECM de presentación aguda en base a la sospecha diagnóstica.
TABLA 4.- Estudios bioquímicos para el diagnóstico de ECM en sangre
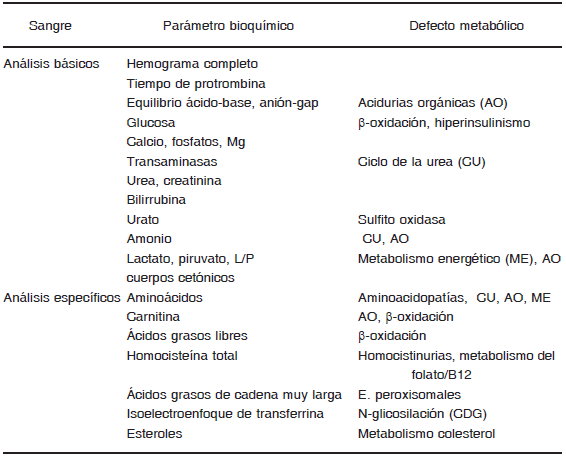
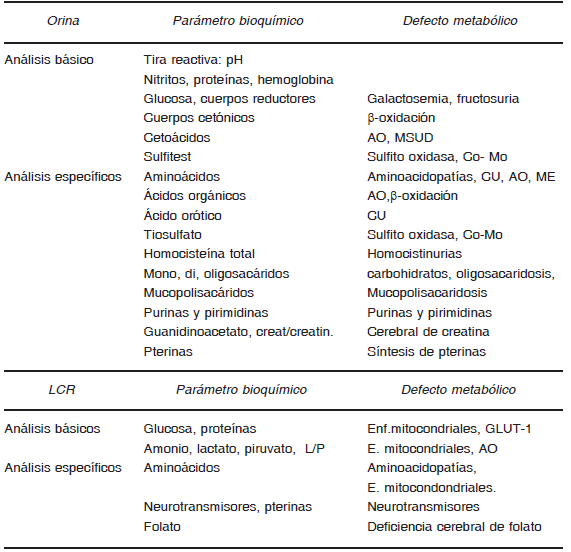
TABLA 5.- Estudios bioquímicos para el diagnóstico de ECM en orina y LCR
Dada la heterogeneidad del conjunto de los ECM hemos considerado para el análisis bioquímico tres grandes grupos1:
- Errores congénitos del metabolismo de moléculas complejas.
- Errores congénitos del metabolismo intermediario o de las moléculas simples.
- Errores congénitos del metabolismo energético.
En el primer grupo se incluyen ECM que implican las moléculas complejas localizadas en el lisosoma, peroxisoma, retículo endoplásmico o aparato de Golgi. Los síntomas clínicos que muestran acostumbran a orientar sobre el origen de dichos ECM, que en algunos casos serán directamente confirmados enzimática y/o genéticamente. No obstante, existen algunos estudios específicos como el análisis de los ácidos grasos de cadena muy larga para el diagnóstico de enfermedades peroxisomales y el isoelectroenfoque de la transferrina para los defectos congénitos de la glicosilación, que permiten seleccionar los pacientes e incluso distinguir los diferentes tipos dentro de estas enfermedades.
El segundo grupo incluye principalmente ECM intermediario, o de las moléculas simples, que dan lugar a un síndrome de intoxicación o a síntomas neurológicos puros, como las aminoacidopatías, acidurias orgánicas, defectos del ciclo de la urea y del metabolismo de los carbohidratos. En ellos, la alteración del metabolismo se traduce generalmente en anomalías de los perfiles bioquímicos en líquidos biológicos (sangre, orina y/o LCR), que orientan sobre las vías metabólicas afectadas y los puntos concretos de bloqueo, lo que permite confirmar los defectos por análisis enzimáticos o de caracterización de las proteínas mutadas y estudios genéticos.
El tercer grupo incluye los ECM del metabolismo energético, que implican defectos de la cadena respiratoria mitocondrial, metabolismo del piruvato, glucogenosis hepáticas (I y III) y defectos de la β-oxidación de los ácidos grasos. Las alteraciones bioquímicas en algunos casos son específicas (β-oxidación), pero en otros casos son simplemente orientativas.
Los especímenes utilizados para el estudio de pacientes con sospecha de ECM son básicamente líquidos biológicos, sangre, orina y, si es necesario, LCR. Las muestras deben tomarse, a ser posible, en la descompensación metabólica y siempre antes del tratamiento. Deben acompañarse de la información clínica sobre la edad, sexo, antecedentes familiares y personales de interés, breve resumen de la historia, dietas especiales, medicación y qué enfermedad o grupo de enfermedades metabólicas sospechamos. Estos datos son indispensables ya que el diagnóstico definitivo no deriva en general de un dato bioquímico aislado, sino de la interpretación del conjunto de datos bioquímicos y clínicos del paciente. Es útil disponer de perfiles de pruebas bioquímicas a solicitar en sangre, orina y LCR, que incluyan las determinaciones que consideremos útiles en el diagnóstico de un ECM, para que los especímenes (que a veces son muestra única) se recojan de la forma adecuada y sean aprovechados al máximo, ya que se trata con frecuencia de neonatos o pacientes en situación crítica. Los tejidos necesarios para la confirmación diagnóstica pueden recogerse en una segunda etapa, cuando la orientación del origen del ECM se haya alcanzado mediante el análisis de líquidos biológicos.
Entre los análisis bioquímicos aplicados para el diagnóstico de un ECM distinguimos unos procedimientos simples que constituyen el análisis básico de alteraciones metabólicas, que aporta una primera orientación sobre la vía afectada (Tabla 4). Ésta se confirma con la ayuda de otros análisis más específicos, que se realizan, siempre que sea posible, en las mismas muestras de sangre, orina o LCR recogidas en descompensación y dependiendo de la sospecha clínica y de los datos aportados en los análisis básicos (Tablas 4 y 5). Si la combinación de los datos clínicos y bioquímicos obtenidos mediante los procedimientos básicos y los específicos ha permitido una orientación definitiva del defecto, se procede a la realización de estudios de confirmación de la proteína afectada por la mutación del DNA que constituye el origen de la enfermedad. Estos estudios de confirmación enzimática o de caracterización de la proteína alterada, pueden realizarse en suero, leucocitos, eritrocitos, fibroblastos cultivados procedentes de biopsia de piel o en tejidos (músculo, hígado, riñón, corazón, etc.). Los estudios genéticos de las mutaciones implicadas en el defecto completarán el estudio. Para ello se extrae DNA de sangre periférica o tejidos del paciente.
En ocasiones no podremos llegar al diagnóstico y el paciente fallece, en estos casos es muy importante disponer de un protocolo para cuando fallece un paciente con sospecha de ECM, que permita alcanzar el diagnóstico post-mortem y dar el adecuado consejo genético a la familia4, 11.
Aporte de la neuroimagen en la sospecha de los ECM
La neuroimagen ha permitido orientar el diagnóstico de muchos ECM y a su vez proporcionar una inestimable ayuda para seguir la evolución natural de la enfermedad y en especial para evaluar los beneficios de algunas terapias. La mayoría de las técnicas de neuroimagen nos ayudan en la sospecha clínica hacia un ECM desde la ecografía craneal transfontanelar, TAC craneal, RM craneal, RM craneal con espectroscopia, RM funcional, TEP, SPECT, sin olvidar la radiología convencional o incluso la arteriografía cerebral que pueden resultar definitivas en el diagnóstico de algunos ECM.
Hay que tener presente que el patrón radiológico de muchos ECM puede ser inespecífico (PKU, leucodistrofia metacromática, enfermedad de Krabbe, adrenoleucodistrofia o leucinosis, por ejemplo) y en contadas ocasiones la imagen será altamente sugestiva de una u otra enfermedad (S. Leigh, A. Glutárica tipo I, déficit PKAN, etc.). Es en este punto cuando el clínico experimentado y observador frente a una anamnesis y clínica sugestivas, una analítica orientativa y una neuroimagen compatible, podrá orientar mejor la sospecha diagnóstica y confirmará el ECM con la técnica disponible más adecuada (plasma, leucocitos, fibroblastos, músculo, hígado, etc.).
Es útil identificar si la afectación es exclusiva de la sustancia blanca o gris; en muchas ocasiones es mixta y en otras inicialmente afecta una de ellas y se extiende al resto del cerebro según la fase evolutiva de la enfermedad1, 12, 13. Lógicamente intervienen otros factores aparte del estadio evolutivo de la enfermedad, como pueden ser los problemas perinatales o postnatales asociados (acidosis, hipoxia, infecciones, hipoglucemia, convulsiones, etc.), las descompensaciones o la terapéutica empleada (leucinosis, déficit biotinidasa, fenilcetonuria, acidosis lácticas, acidurias orgánicas, etc.) con lo que la imagen puede variar. Otro hecho interesante es que algunos pacientes afectos de hiperglicinemia no cetósica, déficit de PDH, E. Menkes o A. Glutárica tipo II pueden manifestar en la neuroimagen lesiones malformativas como agenesia del cuerpo calloso o displasias corticales o malformaciones cerebelosas12, 13.
Uno de los últimos avances en neuroimagen es la RM craneal con espectroscopia (RME). Como la RM convencional, se basa en la propiedad que presentan algunos núcleos atómicos de absorber selectivamente la energía de radiofrecuencia cuando se colocan bajo un campo magnético. Este exceso energético es liberado por los núcleos mediante un proceso de relajación nuclear3. Es una técnica no invasiva que aporta una información bioquímica y permite el estudio de algunos metabolitos del sistema nervioso central12. La RME ofrece pues información bioquímica a diferencia de la RM que ofrece información espacial. Disponemos de experiencia con la determinación del N-Acetil aspartato (NAA), cuya disminución traduce un daño neuronal grave, mientras que su elevación es casi exclusiva de la enfermedad de Canavan14. Por su parte, las elevaciones del lactato en cualquier acidosis láctica pueden evidenciarse a partir de cifras de ácido láctico superiores a 4 mMol/l. Mediante la RME de fósforo-31 e hidrógeno-1 se puede seguir el patrón espectral de diferentes áreas del cerebro tanto de personas sanas como en afectos de encefalopatías metabólicas15, 16. En la actualidad la RME puede ser orientativa en las acidosis lácticas (Fig. 1), E. Canavan, PKU, déficit de creatina cerebral (Fig. 2), etc.12, 13. La disminución del mioinositol apoya fuertemente una hiperamoniemia en sistema nervioso central. Lógicamente la RME nos permitirá en un futuro próximo estudios dinámicos de la vía aerobia y anaerobia, de otras vías metabólicas del estudio y mejor conocimiento de los neurotrasmisores,del metabolismo de los lípidos y en especial mediante la RME hidrogeno-1 del estudio de una serie de compuestos químicos del cerebro con un papel primordial en el funcionalismo neuronal como pueden ser el ácido glutámico, glutamina, GABA, N-acetilaspartato, glicina, colina, glucosa, etc.12.
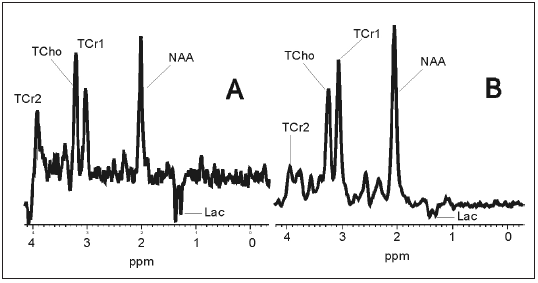
Fig. 1.- Paciente de 18 m con síndrome de Leigh. A) RMs con pico de lactato. B) Después del tratamiento, reducción del lactato.

Fig. 2.- RM corte sagital sin anomalías, en un paciente con defecto del trasportador de creatina cerebral. RMs con práctica ausencia del pico de creatina.
El patrón radiológico puede no ser único para muchas enfermedades; así, por ejemplo, hemos visto un patrón de leucodistrofia en pacientes con síndrome de Leigh, enfermedad de Canavan con afectación del n. pálido, citopatías mitocondriales con/sin elevación lactato mediante RME o incluso calcificaciones de los ganglios basales o áreas de hipoatenuación en la misma localización, aciduria glutárica tipo I con leucodistrofia, con afectación ganglios basales o simplemente con aumento espacios convexidad (Fig. 3) etc. Estos estudios también se están realizando en la actualidad en músculo permitiendo conocer mucho mejor el funcionalismo de algunas vías metabólicas de difícil acceso por otros medios. En otras ocasiones el compromiso es prioritariamente en cerebelo y la orientación diagnóstica será distinta.
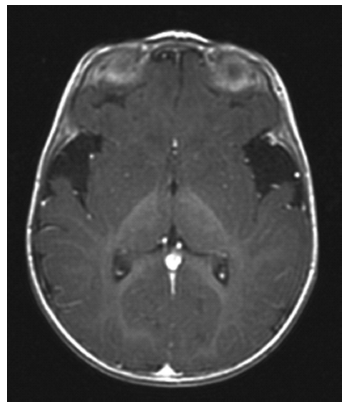
Fig. 3.- RM craneal de un paciente con aciduria glutárica tipo I. Aumento de los espacios de la convexidad en especial a nivel de la cisura de Silvio.
No podemos detallar uno por uno los más de 700 ECM conocidos y analizar en cada uno los hallazgos de neuroimagen, sin embargo existen trabajos que intentan una aproximación a la neuroimagen en los ECM según el patrón de afectación predominante, ya sea de sustancia gris cortical, núcleos subcorticales, sustancia blanca, cerebelo o en la patología malformativa del SNC, para que pueda ser de utilidad en especial para el neuropediatra en su práctica clínica9, 10, 12, 13.
Además existen otras muchas exploraciones complementarias que tienen un importante papel en el diagnóstico de los ECM como pueden ser el registro EEG, los estudios de conducción periféricos, electromiograma, fondo de ojo, electrorretinograma, potenciales evocados visuales y auditivos, entre otros, y que deberemos practicar siempre en base a una hipótesis diagnóstica concreta. Solicitar estos exámenes sin una aproximación clínica-diagnóstica previa generalmente nos podrá conducir a una mayor desorientación para llegar al diagnóstico14, 15.
Es importante además que los pacientes una vez completado el diagnóstico dispongan de una hoja resumen para el caso de una descompensación, con los síntomas de alerta, los análisis a realizar, cuándo debe ingresar el paciente en el Hospital y muy especialmente las medidas de tratamiento en la fase aguda.
El neuropediatra y el pediatra, especialmente en los servicios de Urgencias, debe conocer estos protocolos de actuación y ante la mínima sospecha de un ECM o ante la evidencia clínico/metabólica remitir al paciente a un centro de referencia que disponga de una unidad de metabolopatías. En esta unidad idealmente deberían formar parte un pediatra experto en enfermedades metabólicas, gastroenterólogo, neuropediatra, bioquímico clínico y dietista9, 10.
Una vez confirmado el diagnóstico se deberá proceder a identificar el defecto a nivel enzimático y molecular, iniciar rápidamente un tratamiento dirigido, dar el consejo genético y ofrecer diagnóstico prenatal si lo hubiere.
Conflicto de intereses: Ninguno para declarar.
1. Saudubray JM. Clinical approach to inherited metabolic diseases. In: Fernandes J, Saudubray JM, Van den Berghe G (eds). Springer-Verlag Berlin, 2012. [ Links ]
2. Blom W, Huijmans JGM, van den Berg GB. A clinical biochemist's view of the investigation of suspected inherited metabolic disease. J Inherit Metab Dis 1989; 12: 64-88. [ Links ]
3. Hoffman GF. Selective screening for inborn errors of metabolism: past, present and future. Eur J Pediatr 1994; 153: 52-8. [ Links ]
4. Fernandes J, Saudubray JM. Diagnostic procedures: Function test and postmostem protocol. In: Inborn metabolic diseases. Fernandes J, Saudubray JM, van der Berge G (eds): Springer-Verlag Berlín 1996, p 41-6. [ Links ]
5. Saudubray JM, Charpentier C. Clinical phenotypes: diagnosis/algorithms. In Scriver CR, Beaudet Al, Sly WS, Valle D (ed).The metabolic bases of inherited disease, 8 th ed. New-York: Mc Graw-Hill 2001, p 1327-403. [ Links ]
6. Campistol J. Enfermedades metabólicas de presentación neonatal. Arch Pediatr 1995; 46: 115-7. [ Links ]
7. García-Silva MT. Errores congénitos del metabolismo con repercusión sobre el sistema nervioso en el recién nacido. Cuándo y cómo investigarlos. Rev Neurol 2000; 31: 604-16. [ Links ]
8. Leonard JV, Morris AAM. Inborn errors of metabolism around time of birth. Lancet 2000; 356: 583-7. [ Links ]
9. Aicardi J. Diseases of the nervous system in childhooh. 3rd ed. Londres: Mac Keith Pres, 2009. [ Links ]
10. Sanjurjo, Baldellou. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias 3a ed, Ergon, Madrid, 2010. [ Links ]
11. Marin Valencia I, Vilaseca MA, Thio M, Garcia Cazorla A, Artuch R, Campistol J. Assesment of the perimortem protocol in neonates for the diagnosis of inborn errors of metabolim. Eur J Ped Neurol 2011: 14; 125-30. [ Links ]
12. Campistol J. Aproximación al diagnóstico de los errores congénitos del metabolismo mediante la neuroimagen. Rev Neurol 1999; 28: 16-23. [ Links ]
13. van der Knaap MS, Valk J. Magnetic resonance of myelination and myelin disorders, 3rd. edition Springer-Verlag, Heidelberg 2005. [ Links ]
14. García-Cazorla A, Wolf NI, Serrano M, et al. Inborn errors of metabolism and motor disturbances in children. J Inherit Metab Dis 2009; 32: 618-29. [ Links ]
15. Wolf NI, García-Cazorla A, Hoffmann GF. Epilepsy and inborn errors of metabolism in children. J Inherit Metab Dis 2009; 32: 609-12. [ Links ]
16. Ross B, Michaelis T. Clinical applications of magnetic resonance spectroscopy. Magn Reson Q 1994; 10: 191-247. [ Links ]

