










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680
Medicina (B. Aires) vol.73 supl.1 Ciudad Autónoma de Buenos Aires Sept. 2013
ACTUALIZACIÓN EN NEUROLOGÍA INFANTIL IV
Crisis febriles simples y complejas, epilepsia generalizada con crisis febriles plus, FIRES y nuevos síndromes
Noris Moreno de Flagge
Hospital del Niño, Panamá
Dirección postal: Dra. Noris Moreno de Flagge, Apartado postal 0819-00480, El Dorado, Panamá
e-mail: norismflagge@hotmail.com
Resumen
Las convulsiones febriles representan la mayoría de las convulsiones en el niño. Se ha descrito que 2-5% de los niños experimentan convulsiones febriles antes de los 5 años de edad, aunque en algunas poblaciones se ha descrito hasta un 15%. Es una causa común de admisión en pediatría y de preocupación de los padres. Puede ser la primera manifestación de una epilepsia. Un 13% de pacientes que desarrollan epilepsia tienen antecedente de convulsiones febriles y 30% de estos pacientes se presentan con convulsiones recurrentes. Sus características fenotípicas nos permiten, en su gran mayoría, clasificarlas, tomar una actitud terapéutica y elaborar un pronóstico. Se puede describir un espectro de su gravedad desde las convulsiones febriles simples hasta las más complejas como las convulsiones febriles plus que comprenden los síndromes de Dravet y FIRES. En los últimos años se han hecho descubrimientos importantes que definen su carácter genético, entrelazándose cada vez más con diferentes afecciones de tipo epiléptico que nos obliga a un seguimiento neurológico más estrecho de muchos de estos niños con convulsiones febriles. Hacemos una revisión bibliográfica con el objetivo de actualizar los conocimientos sobre las convulsiones febriles, su pronóstico y su relación con los nuevos síndromes epilépticos.
Palabras clave: Convulsión febril; Nuevos síndromes; Dravet; FIRES; Genética
Abstract
Simple febrile seizure, complex seizure, generalized epilepsy with febriles seizure plus, FIRES and new syndromes. Febrile seizures are the most common seizures in childhood. They have been observed in 2-5% of children before the age of 5, but in some populations this figure may increase to 15%. It is a common cause of pediatric hospital admissions and cause of anxiety for parents. Febrile seizures could be the first manifestation of epilepsy. About 13% of epileptic patients have a history of febrile seizure, and 30% have had recurrent febrile seizures. Their phenotypic characteristics allow, in the majority of cases, a classification of the seizure, an elaboration of a prognosis and to assume a specific therapeutic attitude. It is possible to describe a spectrum according to their severity, from the benign simple seizure to the more complex, febrile seizure plus, Dravet`syndrome, and FIRES. During the past decade, molecular genetic studies have contributed to the identification of genetic factors involved in febrile seizure and related disorders, making the necessity of a careful follow up of these patients in order to detect risk factors earlier. We have reviewed the medical literature to update current knowledge of febrile seizures, their prognosis and their relation to new epileptic syndromes.
Key words: Febrile seizure; New syndromes; Dravet´syndrome; FIRES; Genetic
Las convulsiones febriles (CF) son muy frecuentes y representan la mayoría de las convulsiones en el niño. Se ha descrito que 2-5% de los niños experimentan convulsiones febriles antes de los 5 años de edad, aunque en algunas poblaciones se ha descrito hasta un 15%. Es una causa común de admisión en pediatría y de preocupación de los padres. Es el prototipo de una crisis epiléptica, y sin embargo no se considera como tal por su factor provocador, la fiebre. Aunque fue descrita ya desde el tiempo de los primeros griegos (Hipócrates 440 años a.c.), no fue hasta esta centuria que las convulsiones febriles se reconocieron como un síndrome diferente separado de la epilepsia.
Estudios genéticos extensos han demostrado que hay por lo menos nueve genes involucrados en la génesis de las convulsiones febriles y algunas formas comunes de ellas son desórdenes genéticos complejos que se piensa son influenciados por variaciones en varios genes susceptibles.
Definición
Hay dos definiciones aceptadas de convulsiones febriles. Una propuesta por The National Institute of Health (NIH), en 1980, la definió como " un evento en la infancia o niñez que ocurre usualmente entre los 3 meses a 5 años de edad, asociado a fiebre pero sin ninguna evidencia de infección intracraneal o causa definida para la convulsión". Esta definición excluye convulsiones febriles en niños que han tenido convulsiones afebriles previas. Por otro lado, la Liga Internacional contra la Epilepsia (ILAE) en 1993 definió la CF como: "una crisis que ocurre en el niño desde el primer mes de edad, asociada a enfermedad febril no causada por una infección del SNC, sin crisis neonatales previas o crisis epiléptica previa no provocada, y no reuniendo criterios para otro tipo de crisis aguda sintomática"1, 2. La primera es la más conocida y la segunda más utilizada por los epileptólogos. Ambas toman en consideración tres componentes críticos en la definición: edad, convulsión y fiebre. Difieren, sin embargo, en la edad de presentación, siendo para la NIH a los 3 meses y para la ILAE en el primer mes. Esta última no define una edad final, lo que para la NIH es a los 5 años. No excluyen niños con daño neurológico previo y ninguna de las dos define la temperatura durante la convulsión febril, ni describen la convulsión.
No se define la temperatura menor necesaria para la CF, lo que dificulta el diagnóstico en ocasiones3. Esto obligó a la Academia Americana de Pediatría (1999), a elaborar parámetros para definir las convulsiones febriles simples estrictamente, como aquellas que ocurren en niños previamente sanos, con edades que oscilan entre 6 meses a 5 años de edad cuyas convulsiones son breves (menos de 15 minutos), generalizadas y ocurren sólo una vez en un período de 24 horas durante la fiebre. Se excluyen niños cuyas convulsiones se atribuyen a infección del SNC o aquellos que han tenido una convulsión afebril previamente o tienen una anormalidad del SNC4. Sin embargo, el criterio aceptado en general para una CF incluye:
- Una convulsión asociada a una temperatura elevada de más de 38 °C
- En un niño menor de 6 años
- No signos de infección o inflamación del SNC
- No anormalidades metabólicas agudas que puedan producir convulsiones
- No historia de convulsiones afebriles previas
Epidemiología
Temperatura
¿Cuál es la relación de la fiebre y la convulsión febril? Numerosos estudios epidemiológicos definen la misma como > 38 °C y otras > 38.4 °C5-7. Las convulsiones febriles pueden ocurrir antes de que la fiebre sea aparente o tarde o temprano en el curso de una enfermedad febril4.
Edad
La mayoría de las CF ocurre entre 6 meses y 3 años de edad con un pico de incidencia a los 18 meses de edad6-9; 6-15% ocurre después de los 4 años y es muy raro que se inicien después de los 6 años. Otros autores consideran que el rango está entre los 6 meses a 5 años, y si se inician fuera de ese rango de edad no se deben considerar convulsiones febriles simples8.
Sexo
Los niños están afectados ligeramente más que las niñas en un 60%.
Prevalencia
La prevalencia es aproximadamente 3%, pero es más alta en ciertos grupos étnicos como Japón (7%) y Guam (14%). La incidencia anual es de 460 por 100 000 en la población de niños de 0-4 años (8-10).
Convulsión
El Consenso de la NIH la describe como un evento y no usa la palabra convulsión10, 11. Sin embargo, se considera a las convulsiones tónico-clónicas (80%) como las típicas de una convulsión febril y el prototipo de una convulsión epiléptica. Tónicas (13%), atónicas (3%) y crisis tónico-clónicas de inicio focal o unilateral (4%). Las sacudidas mioclónicas o ausencias no se consideran parte de una convulsión febril. Las crisis se pueden repetir en la misma enfermedad febril en un 16%.
Etiología y patogénesis
No se conoce cómo y por qué las convulsiones se generan en respuesta a la fiebre, puede ser que factores inducidos por la fiebre (ej.: interleukina-1 beta), son pro-convulsivos en individuos que son susceptibles basados en el estado de desarrollo del cerebro y su susceptibilidad genética12-14.Ciertos canales de sodio en el cerebro son sensibles a la temperatura y pueden generar una actividad neuronal sincronizada asociada a la fiebre15, 16.También hay evidencia que sugiere que la hiperventilación y alcalosis inducida por la hipertermia puede jugar un papel17. Las CF pueden ocurrir tanto con infecciones virales como bacterianas. Aunque las infecciones virales son las más comunes, tal vez por la mayor incidencia de las mismas en niños, las causas varían y pueden incluir infecciones del tracto respiratorio alto o faringitis (38%), otitis media (23%), neumonía (15%), gastroenteritis (7%), roséola infantil (5%) e infecciones no virales (12%). Se ha postulado también una asociación específica entre el herpes virus humano 6 (roséola infantil) y las CF18.
Inmunizaciones
El riego de CF aumenta después de vacunación con difteria (toxoide tetánico con células completas de pertusis (DPT), y con vacunas de sarampión, parotiditis y rubéola (MMR).
Factores predisponentes
La susceptibilidad a las CF se ha asociado a anormalidades en los neurotransmisores.
Factores inmunológicos
Como la concentración de neopterina puede estar elevada en el LCR de niños con CF, el cual es secretado y activado por los macrófagos. Esto sugiere un mecanismo inmunológico en el SNC como consecuencia de una invasión viral o bacteriana al mismo. La deficiencia de hierro también se ha asociado a las causas de CF.
Bases genéticas
La predisposición genética es importante, considerando que las convulsiones febriles a menudo son familiares19, 20. Los niños con hermanos o padres con convulsiones febriles tienen un riesgo 4 o 5 veces mayor que la población general de presentar convulsiones febriles. Se ha comunicado además, que la tasa de concordancia en gemelos homocigóticos y dicigóticos es de 70 y 20% respectivamente. El modo de herencia es desconocido aunque se ha sugerido que es poligenética en pacientes aislados y autosómica dominante con penetrancia incompleta en familias con CF recurrentes. Se ha demostrado una heterogeneidad genética con estudios de ligamentos de familias grandes con herencia autosómica dominante incompleta, permitiendo identificar cinco diferentes locus vinculados a la CF. Estos se encuentran mapeados en los cromosomas: 2, 5, 6, 8 y 19. Se está intentando correlacionar cómo esta determinación genética pudiera contribuir al posterior desencadenamiento de otras crisis epilépticas que aparecen tardíamente en niños con CF. Aunque no se ha establecido un gen o locus para CF, estudios clínicos y de genética molecular sugieren que son numerosos los genes síndrome-específicos para las crisis febriles.
Uno de los síndromes genéticos más importantes en los que se ha identificado un defecto genético lo constituye el síndrome Convulsiones Generalizadas con Crisis Febriles Plus (epilepsia autosómica dominante con convulsiones febriles plus) (GEFS+) caracterizado por un fenotipo heterogéneo de convulsiones epilépticas focales y generalizadas, a partir del cual se está abriendo camino para el descubrimiento de nuevos síndromes21. Las evidencias sugieren el compromiso de varios canales de sodio, receptores GABA y proteínas auxiliares, en la patogénesis de las CF+ y aun en las CF simples19.
Diagnóstico diferencial
Hay que diferenciar las CF de otros eventos asociados a fiebre como los síncopes, rubores, convulsiones anóxicas, espasmos del sollozo, pérdida de la conciencia y apnea, que puede presentarse en enfermedades febriles y se pueden descartar con una historia clínica cuidadosa. La sociedad de pediatría británica considera necesario diferenciar convulsión febril de convulsión con fiebre. Entre las convulsiones con fiebre se reconocen las meningitis piógenas o virales, la encefalitis herpética, otras encefalitis agudas, la parálisis cerebral con infecciones intercurrentes, las enfermedades metabólicas o degenerativas con convulsiones desencadenadas por la fiebre11.
Hay una entidad más frecuentemente descrita, primero en Asia y luego en países de América y Europa; se caracteriza por crisis convulsivas afebriles en niños con gastroenteritis y temperatura menor de 38 °C. Una serie de casos demostró que estos niños presentaban convulsiones repetitivas, en racimos, generalizadas o focales, con o sin fiebre por varios días y durante una gastroenteritis leve. Los niños evolucionaban sin recurrencia de crisis y sin deterioro del desarrollo psicomotor. En nuestro hospital pudimos evaluar, recientemente, (año 2012) a 10 niños con características similares, muchos de ellos ameritando observación en cuidados intensivos, pero con una supervivencia de 100% y sin deterioro de su desarrollo. Comparados con los niños con convulsiones febriles, estos niños tienen una incidencia de recurrencia menor, aunque las crisis son más repetidas y en racimos durante el proceso agudo22.
Clasificación
Las CF se dividen en dos categorías: simples (benignas) y complejas, basadas en las características clínicas. Las CF simples son las más comunes (70% de todas) y se caracterizan por convulsiones que duran menos de 15 minutos, no tienen carácter focal, no repiten en las siguientes 24 horas y se resuelven espontáneamente. Las CF complejas se caracterizan por episodios que duran más de 15 minutos, tienen carácter focal o parálisis postictal, repiten en las siguientes 24 horas y si ocurren en serie la duración total es de más de 30 minutos1, 2, 23, 24. Las CFs complejas ocurren con un amplio margen de 9-35%, debiéndose esto probablemente a la dificultad de diferenciar muchas veces una crisis simple de una compleja y tal vez una febril de una afebril. Aunque es fácil diferenciar una crisis compleja prolongada o repetitiva, una focal o con manifestaciones sutiles de ausencia o asimetrías motoras puede pasar muchas veces desapercibida24.
Estado convulsivo febril: Se considera estado convulsivo febril (ECF) a una convulsión o a una serie de convulsiones con fiebre sin recuperar conciencia entre ellas, durante un período de tiempo de 30 minutos o más. Está considerado como una CF compleja, por su duración de 30 minutos o más sin recuperar la conciencia. Representa un 25% de todos los status convulsivos en el niño25. Sin embargo, aunque las CFs complejas tengan una incidencia baja, el estatus convulsivo febril representa un 25% de todos los estados convulsivos en el niño. El estudio multicéntrico de la Academia Americana de Neurología, (FEBSTAT) demostró que los virus herpes humano (HHV) 6-B y HHV 7-(virus de roséola) son responsables de un tercio de los estados convulsivos febriles en el niño26.
Fenotipos clínicos
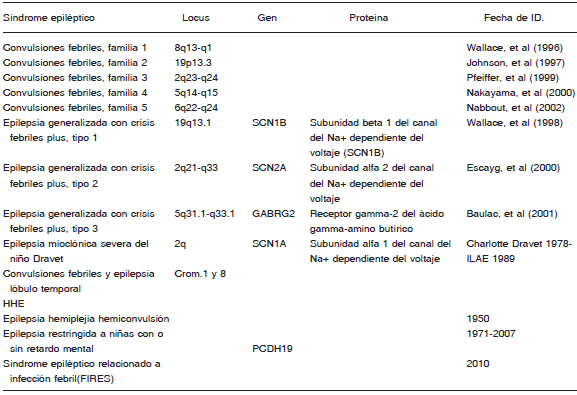
Los avances en la biología molecular y la identificación de síndromes epilépticos relacionados a las CF han permitido definir varios fenotipos clínicos de CF que constituyen un continuo neurobiológico desde la CF simple única en un extremo y las CF complejas que constituyen manifestaciones iniciales del síndrome de Dravet27 (Tabla 1).
TABLA 1.- Fenotipos clínicos de CF

Convulsión febril plus (CF+)
Tiene una gran tendencia familiar. El término CF+ o atípicas, se usa para referirse a las CF que comienzan muy temprano, antes de los 6 meses de edad, a diferencia de las CF clásicas y que persisten después de los 6 años, pero tienden a desaparecer alrededor de los 11 años, pero si persisten pueden ser de difícil control. Usualmente se repiten con frecuencia y los niños con CF+ pueden también presentar convulsiones afebriles9. Una CF compleja, prolongada o focalizada, o CF+ después de los 5 años, puede evolucionar hacia una epilepsia de lóbulo temporal o hacia un síndrome de Dravet años después, o puede sencillamente desaparecer sin dejar secuelas28, 29. En su descripción original, Scheffer y Berkovic definieron como CF+ aquellas que ocurrían en los niños después de los 6 años y que podían ocurrir luego sin fiebre, por lo tanto epilépticas30. De acuerdo a Berkovic, la clave para diagnosticar el fenotipo de las CF+ estriba en la continuidad de las convulsiones generalizadas después de los 5 años, y no tanto la presencia o no de fiebre. En algunos niños con CF+, las primeras convulsiones con fiebre ocurrieron después de los 6 años y en otros grupos con CF todas aquellas convulsiones después de los 6 años ocurrieron sin fiebre.
Epilepsia generalizada y crisis febriles plus (EGCF+)
Nace así el concepto de epilepsia generalizada con convulsiones febriles plus (EGCF+), cuando en 1997 Scheffer y Berkovic describieron paralelamente un nuevo tipo de convulsiones relacionadas a fiebre en una numerosa familia australiana30. Describieron numerosos miembros que presentaban convulsiones de tipo epiléptico en diferentes generaciones. Lo que encontraron fue que la mayoría comenzaban con CF las cuales no paraban a la edad usual, sino que persistían más allá de los 6 años, o presentaban crisis afebriles, que podía ocurrir luego de un período libre de crisis. Además describieron estas CF+, las cuales podían variar de una persona a otra, es decir con diferentes fenotipos. El fenotipo más común es el de CF a menudo asociado a convulsiones afebriles con crisis tipo tónico clónicas generalizadas; en un tercio, pueden también ser de tipo ausencias, mioclónicas, crisis atónicas, conformando un espectro muy amplio, con factores clínicos y genéticos comunes, pero con muy diferente pronóstico. Más adelante Abou-Kahlil31 lo describe con crisis parciales, asociado a epilepsia de lóbulo temporal y luego Scheffer a epilepsia de lóbulo frontal32. Esto ha revolucionado diferentes conceptos en las epilepsias. Es un cuadro genético de tipo autosómico dominante con penetrancia incompleta, en la que intervienen mutaciones de los genes que codifican los canales iónicos de sodio dependientes del voltaje subunidad alfa1 (SCN1A), alfa2(SCN2A) y B1(SCN1B) y la subunidad gamma 2 del receptor GABA(GABRG2)33 (Tabla1).
Es de mucha importancia clínica ya que relaciona las CF con diferentes síndromes epilépticos y documenta la relación genética entre ellos ya sea benigna o grave, focal o generalizada. No se ha definido su prevalencia aún. La ILAE (Liga Internacional contra la Epilepsia) la ha incluido en la clasificación de la epilepsia, como un síndrome en desarrollo34.
Manifestaciones clínicas de EGCF+
Las manifestaciones son variables de un paciente a otro. El espectro es extenso desde las formas familiares leves, CF clásicas que desaparecen antes de los 6 años hasta las más graves o síndrome de Dravet. Dentro de la misma familia el cuadro es variable, pero generalmente benigno.El fenotipo más común incluye CF y CF+, en el cual las CF persisten más allá de los 6 años de edad y están asociadas a crisis afebriles, la mayoría de las veces generalizada, y más raras veces parciales. La coexistencia de diferentes tipos de convulsiones epilépticas en el mismo paciente orienta hacia la constitución de síndromes epilépticos específicos más graves como la epilepsia mioclónica astática, la epilepsia mioclónica juvenil, algunas con alteraciones electroencefalografías que no corresponden a ningún síndrome ya establecido en la clasificación de las epilepsias. Además la epilepsia hemiconvulsiva, epilepsia de lóbulo temporal y epilepsia de lóbulo frontal, extendiendo la clasificación de la EGCF+ al incluir las convulsiones parciales, ampliando así el fenotipo de la misma (Tabla 2).
TABLA 2.- Fenotipo de CF y expresión genética
Evolución y tratamiento de EGCF+
La evolución así como la respuesta al tratamiento es también variable dentro de la misma familia. Las crisis afebriles pueden ser poco frecuentes y desaparecer luego de algunos años sin dejar secuelas, y otras veces evolucionar hacia una forma muy grave de epilepsia. La respuesta al tratamiento es también variable, algunos responden bien a la monoterapia, mientras que otros pueden ser resistentes a los anticonvulsivantes. Algunos pacientes pueden desarrollar retraso mental.
Síndrome de Dravet
Conocida también como epilepsia mioclónica grave (SMEI): descrito por Charlotte Dravet en 197835. Es una encefalopatía, considerada la más grave de las epilepsias relacionadas a la mutación SCN1A. Incluida en la clasificación de la epilepsia de la ILAE en 1989 como un desorden criptogénico con características focales y generalizadas. En la revisión de 2008 la ILAE la clasifica como una encefalopatía de inicio en el primer año de la vida con bases genéticas, más frecuentemente una canalopatía y posiblemente parte del espectro de la EGCF+, ya que el gen SCN1A que codifica para la subunidad alfa 1 del canal de sodio neuronal se encuentra con mutaciones hasta en un 80% de los pacientes con epilepsia mioclónica grave de la infancia35. En dicha revisión es clasificada además como un síndrome epiléptico, genético en desarrollo.
Cuadro clínico: Se inicia entre los 4 a 10 meses de edad como CF+ frecuentes, prolongadas y a veces unilaterales, que se producen con temperaturas no muy altas y que tienden a recurrir en los dos primeros años, cada 4-6 semanas . A partir de los 18 -24 meses aparece las crisis afebriles, inicialmente tónico-clónicas y luego con crisis mioclónicas y ausencias atípicas y estatus epiléptico no convulsivo. En un mismo paciente pueden coexistir más de un tipo de convulsiones. La vacunación y enfermedades virales banales pueden ser factores desencadenantes36. En la mayoría de los pacientes se puede observar deterioro del desarrollo psicomotor en diferentes grados. Con el tiempo pueden aparecer otros síntomas neurológicos como ataxia o temblores. El electroencefalograma (EEG) es inicialmente normal y luego aparecen anormalidades como disrritmia de ondas lentas con espigas difusas y focales.
Tratamiento: Es un síndrome epiléptico refractario a tratamiento. Se deben evitar los anticonvulsivantes dependientes de los canales del sodio, como carbamazepina y lamotrigina. Parecen ayudar en muchos casos el ácido valproico, stiripentol, topiramato y la dieta cetógena29.
Sindrome de Juberg y Hellman o epilepsia restringida en niñas con o sin retardo mental (EFMR)
Es un nuevo síndrome epiléptico descrito en mujeres y relacionado a fiebre, considerado dentro de las canalopatías; se asocia al gen PCDH19. Fue descrito por primera vez por Juberg y Hellman en 1971 y publicado por primera vez en 200737. Aún no tiene nombre asignado según la clasificación de la ILAE.
Cuadro clínico: se describe en niñas sanas que inician las convulsiones alrededor de 3 a 36 meses (antes de los 3 años). La mayoría de las veces asociado a un cuadro febril y en ocasiones relacionado a cuadro postvacunal. Lo característico es que se presentan en forma agrupada y pueden tener un fenotipo variable con crisis tónico-clónica, parcial, mioclónicas, ausencias, crisis atónicas e incluso se ha descrito estatus hemiclónico. Puede desaparecer a los 12 años, pero la gran mayoría evoluciona hacia crisis crónicas con deterioro intelectual. Los hallazgos electroencefalográficos son variables, desde normal en la primera infancia y luego con enlentecimiento delta difuso o espigas o poliespigas difusas. No se ha determinado un patrón electroencefalográfico con la edad. No se ha descrito anormalidades en la resonancia magnética cerebral (RM) o la tomografía cerebral (TC).
Patrón de herencia atípico. Dominante ligado al cromosoma X. El hombre es el portador y las mujeres presentan la enfermedad.
Se desarrolla por un mal funcionamiento de la Proteína PCDH19 (protocadherin 19). Las cadherinas de las que la PCDH es una estructura similar son proteínas de adhesión celular en este caso dependientes del calcio. En los casos de síndrome de Dravet sin mutaciones en el SCN1A, se debe investigar por el gen Protocadherina 19 (PCDH19) especialmente en mujeres38, 39. Puede ocurrir en niñas normales, aunque se ha descrito en hombres. Dos tercios presentan problemas en su desarrollo: problemas de aprendizaje, retraso mental, depresión y/o problemas como autismo, comportamientos agresivos. Tratamiento: se describe resistencia a los anticonvulsivantes, aunque suele responder a las benzodiacepinas.
Síndrome epiléptico de hemiconvulsión hemiplejía
Fue descrito hace cinco décadas y aún se desconoce su fisiopatología40. Es un síndrome poco común caracterizado por crisis convulsivas hemigeneralizadas o focales de larga duración en forma de estatus epiléptico seguido de parálisis de Todd o hemiplejía permanente. Estas convulsiones focales y prolongadas ocurren durante el curso de una enfermedad febril. Se acompaña de manifestaciones radiológicas de edema citotóxico agudo con atrofia posterior del hemisferio afectado.
Cuadro clínico: los niños usualmente tienen menos de 4 años cuando se presenta el cuadro y cursan una enfermedad febril. Se caracteriza por convulsiones prolongadas de más de 24 horas, tipo clónicas unilaterales que pueden pasar desapercibidas inicialmente. Las sacudidas pueden ser de localización variable y si se prolongan pueden ser bilaterales. Puede haber síntomas autonómicos graves como hipersalivación, trastorno respiratorio con cianosis. Tratamiento: inicialmente de sostén y muchos niños evolucionan bien si se logra controlar el estatus convulsivo inicial. Cuando el edema es grave se ha propuesto el uso de agentes antiedema tipo receptores antagonistas de glutamato NMDA. Después de un período variable de control, meses a años, sin convulsiones, aproximadamente dos tercios de los pacientes desarrollan epilepsia grave refractaria a tratamiento41.
Síndrome epiléptico relacionado a infección febril (FIRES)
El síndrome epiléptico relacionado a infección febril (FIRES) es un síndrome epiléptico cada vez más conocido. Se presenta en niños previamente sanos, como una encefalitis con estatus epiléptico multifocal y evoluciona hacia una epilepsia crónica, refractaria, focal, con deterioro del área cognitiva y comportamental. Fue descripta por primera vez por Awaya y Fukuyama42. Desde entonces ha habido variadas publicaciones43-45 concluyendo todas que no hay agente etiológico, infeccioso, metabólico o genético identificado, y que es resistente a cualquier tratamiento, incluyendo inmunoglobulina, corticoides, plasmaféresis y rituximab46.
Cuadro clínico: La fase aguda está bien descrita en la literatura47. Niños previamente sanos se presentan con una enfermedad de apariencia encefalitis, con convulsiones que rápidamente pasan a un estatus convulsivo refractario requiriendo cuidados en terapia intensiva. Tiene tres fases:1-fase inicial con una convulsión febril simple; 2-pocos días después fase aguda caracterizada por convulsiones recurrentes o estatus epiléptico refractario, a menudo sin fiebre y generalmente sin compromiso neurológico adicional (clásico fenotipo puro de convulsión); 3-por último fase crónica, epilepsia resistente a tratamiento y compromiso neuropsicológico grave.
El diagnóstico diferencial es con una encefalitis viral, una encefalomielitis, encefalitis de Rasmussen, anormalidades estructurales cerebrales y trastornos metabólicos.
Etiología: Todos los nombres se refieren a una etiología infecciosa relacionada con énfasis en una presentación aguda. El inicio con la presencia de una enfermedad febril en la mayoría de los pacientes con una pleocitosis ligera en el LCR lleva a pensar en una infección viral o una inflamación con un componente inmunológico. Sin embargo, no se ha aislado ningún patógeno y no hay evidencia histológica de inflamación en las autopsias. Se ha relacionado con el síndrome de Dravet por su presentación catastrófica en niños previamente sanos, donde un cuadro genético de tipo canalopatía del canal de sodio es desenmascarado por una enfermedad febril o después de vacunación, con estatus epiléptico pasando a una epilepsia crónica refractaria al tratamiento y regresión del desarrollo. Sin embargo, estudios recientes demuestran que el FIRES no pertenece al grupo de las canalopatías48. Continúa siendo una enfermedad misteriosa en espera de descubrirse su etiología.
Las convulsiones febriles, aunque benignas la mayoría de las veces, pueden evolucionar hacia crisis de muy difícil manejo, afectando la calidad de vida de los niños. Aun así poco se conoce de los mecanismos generadores de muchas de estas enfermedades de origen febril. Partiendo de las convulsiones febriles simples y complejas hasta las más estudiadas últimamente, convulsiones febriles plus + (atípicas) y la Epilepsia Generalizada con convulsiones febriles +, se han descripto síndromes epilépticos que atraen el interés de los especialistas en neurología y epilepsia con miras a una intervención temprana, como son el síndrome de Dravet y evitar los efectos devastadores de muchos síndromes epilépticos nuevos. El rápido crecimiento en los avances de estudios genéticos ha contribuido en los últimos años a una mejor comprensión de algunos de estos síndromes epilépticos relacionados a fiebre; sin embargo, aún no se puede hacer una correlación clara entre el fenotipo y una alteración genética definida debido a la complejidad de los genes y sus mutaciones dentro de una misma familia, creando diferentes síndromes. Creemos que es en la vía del conocimiento de los estudios genéticos, inmunológicos y metabólicos que está nuestra esperanza de aclarar la mayoría de estos síndromes epilépticos.
Conflicto de intereses: Ninguno para declarar.
1. Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989; 30: 389-99. [ Links ]
2. Engel J Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001; 42: 796-803. [ Links ]
3. Waruiru R, Appleton R. Febrile seizure: an Update. Arch Dis Child 2004; 89: 751-6. [ Links ]
4. American Academy of Pediatrics. Committee on Quality Improvement, Subcommittee on Febrile Seizures. Practice parameter: long-term treatment of the child with simple febrile seizures. Pediatrics 1999; 103: 1307-9. [ Links ]
5. Annegers JF, Hauser WA, Shirts SB, et al. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med 1987; 316: 493-8. [ Links ]
6. Hauser WA. The prevalence and incidence of convulsive disorders in children. Epilepsia 1994; 35(suppl 2): S1-6. [ Links ]
7. Offringa M, Hazebroek-Kampschreur AAJM, Derksen-Lubsen G. Prevalence of febrile seizures in Dutch school children. Pediatr Perinat Epidemiol 1991; 5: 181-8. [ Links ]
8. Wallace SJ. The Child with Febrile Seizures. London: Butterworth, 1988. [ Links ]
9. Panayiotopoulos CP. The Epilepsies: Seizures, Syndromes and Management. Oxfordshire UK: Bladon Medical Publishing, 2005. [ Links ]
10. Forsgren L, Sidenvall R, Blomquist HK, et al. A prospective incidence study of febrile convulsions. Acta Pediatr Scand 1990; 79: 550-7. [ Links ]
11. Wallace SJ. Febrile seizures. In: Wallace SJ, Farrell K, (eds). Epilepsy in children. London: Arnold 2004, p 123-30. [ Links ]
12. Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med 1976; 295:1029-33. [ Links ]
13. Nakayama J. Progress in searching for the febrile seizure susceptibility genes. Brain Dev 2009; 31: 359. [ Links ]
14. Heida JG, Moshé SL, Pittman QJ. The role of interleukin-1beta in febrile seizures. Brain Dev 2009; 31: 388. [ Links ]
15. Shibasaki K, Suzuki M, Mizuno A, et al. Effects of body temperature on neural activity in the hippocampus: regulation of resting membrane potentials by transient receptor potential vanilloid 4. J Neurosci 2007; 27: 1566. [ Links ]
16. Thomas EA, Hawkins RJ, Richards KL, et al. Heat opens axon initial segment sodium channels: a febrile seizure mechanism? Ann Neurol 2009; 66: 219. [ Links ]
17. Schuchmann S, Schmitz D, Rivera C, et al. Experimental febrile seizures are precipitated by a hyperthermia-induced respiratory alkalosis. Nat Med 2006; 12: 817. [ Links ]
18. Barone SR, Kaplan MH, Krilov LR. Human herpesvirus-6 infection in children with first febrile seizures. J Pediatr 1995; 127: 95-7. [ Links ]
19. Hirose S, Mohney RP, Okada M, et al. The genetics of febrile seizures and related epilepsy syndromes. Brain Dev 2003; 25: 304-12. [ Links ]
20. Berkovic SF, Scheffer IE. Genetics of the epilepsies. Epilepsia 2001; 42(Suppl 5):16-23. 11887962 [ Links ]
21. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997; 120: 479-90. [ Links ]
22. Uemura N, Okumura A, Negoro T, et al. Clinical features of benign convulsions with mild gastroenteritis. Brain Dev 2002; 24: 745-9. [ Links ]
23. Fishman MA. Febrile seizures. In: Principles and Practice of Pediatrics, 3rd. ed., McMillan JA, et al. (eds), JB Lippincott, Philadelphia 1999, p 1949. [ Links ]
24. Nelson KB, Ellenberg JH. Febrile Seizures (eds), Raven Press, New York, 1981. [ Links ]
25. Berg AT, Steinschneider M, Kang H, et al. Classification of complex features of febrile seizures: interrater agreement. Epilepsia 1992; 33: 661-6. [ Links ]
26. Epstein LG, Shinnar S, Hesdorffer DC, et al. Human herpesvirus 6 and 7 in febrile status epilepticus: The FEBSTAT study. Epilepsia 2012; 53: 1481-8. [ Links ]
27. Maytal J, Shinnar S. Febrile status epilepticus. Pediatrics 1990; 86: 611-6. [ Links ]
28. Nieto Barrera M, NietoJimenez M, Nieto Jimenez E. Edita: Sociedad Española de Medicina Extrahospitalaria y Atención primaria (SEPEAP), Editorial: Ediciones Ergón S.A, Pediatría Integral 2007; XI(9), p 753-68. [ Links ]
29. Caraballo R, Fejerman N. Tratamiento de las epilepsias, 1a ed. Buenos Aires: Editorial Médica Panamericana, 2009. [ Links ]
30. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997; 120: 479-90. 912605. [ Links ]
31. Abou-Khalil B, Ge Q, Desai R, et al. Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology 2001; 57: 2265-72. [ Links ]
32. Scheffer IE, Harkin LA, Dibbens LM, et al. Neonatal epilepsy syndromes and generalized epilepsy with febrile seizures plus (GEFS+). Epilepsia 2005; 46: 41-7. [ Links ]
33. Baulac S, Gourfinkel-An I, Nabbout R. Fever, genes and epilepsy. Lancet Neurol 2004; 3: 421-30. [ Links ]
34. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1989; 30: 389-99. [ Links ]
35. Marine C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007; 48: 1678-96. [ Links ]
36. Dravet C. Severe myoclonic epilepsy of infancy. In Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P (eds). Epileptic syndromes in infancy, childhood and adolescence, 4th ed. John Libbey Euro text Ltd Montrouge, 2005, p 89-115. [ Links ]
37. Scheffer IE, Turner SJ, Dibbens LM, et al. Epilepsy and mental retardation limited to female: an under recognized disorder. Brain 2007; 131: 918-27. [ Links ]
38. Dibbens LM, Kneen R, Bayly MA, et al. Recurrence risk of epilepsy and mental retardation in females due to parental mosaicism of PCDH19 mutations. Neurology 2011; 76: 1514-9. [ Links ]
39. Marini C, Meid D, Parmeggiani L, et al. Infantile onset focal epilepsy and epileptic encephalopathies associated with PCDH19 gene mutations: New de novo and familial mutations. Epilepsia 2010; 51(Suppl. 4): 1-89. [ Links ]
40. Tenney J, Schapiro MB. Child neurolgy: hemiconvulsion-hemiplegia-epilepsy syndrome. Neurology 2012; 79: e1-4. doi. [ Links ]
41. Auvin S, Devisme L, Maurage C, et al. Neuropathological and MRI finding in an acute presentation of hemiconvultion-hemiplegia: a report with pathophysiological implication. Seizure 2007; 16: 371-6. [ Links ]
42. Awaya Y, Fukuyama Y. Epilepsy sequelae of acute encephalitis or encephalopathy (3rd report). Jpn J Psychiatry Neuro 1986; 40: 385-7. [ Links ]
43. Baxter P, Clarke A, Cross H. Harding B, et al. Idiophatic catastrophic epileptic encephalopathy presenting with acute onset intractable status. Seizure 2003; 12: 379-87. [ Links ]
44. Sakuma H, Awaya Y, Shiomi M, et al. Acute encephalitis with refractory, repetitive parcial seizure (AERRPS): a peculiar form of childhood encephalitis. Acta Neurol Scand 2010; 121: 251-6. [ Links ]
45. Nabbout R, Vezzani A, Dulac O, et al. Acute encephalitis with inflammation-mediated status epileptic. Lancet Neurol 2011; 10: 99-108. [ Links ]
46. van Baalen A, Häusler M, Boor R, et al. Febrile infection-related epilepsy syndrome (FIRES): A nonencephalitic encephalopathy in childhood. Epilepsia 2010; 51: 1323-8. [ Links ]
47. Howell K, Katanyuwong K, Mackay M, et al. Long-term follow-up of febrile infection-related epilepsy syndrome. Epilepsia 2012; 53: 101-10. [ Links ]
48. Carranza Rojo D, Harvey AS, Iona X, et al. Febrile infection-related epilepsy syndrome is not caused by SCN1A mutations. Epilepsy Res 2012; 100:194-8. [ Links ]

