










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680
Medicina (B. Aires) vol.74 no.2 Ciudad Autónoma de Buenos Aires abr. 2014
ARTÍCULO ESPECIAL
Funciones de los canales iónicos CFTR y ENAC en la fibrosis quística
Alejandra G. Palma, Basilio A. Kotsias, Gabriela I. Marino
Laboratorio de Canales Iónicos, Instituto de Investigaciones Médicas Alfredo Lanari, IDIM-CONICET, Universidad de Buenos Aires, Buenos Aires, Argentina
Dirección Postal: Gabriela I. Marino, Instituto de Investigaciones Médicas Alfredo Lanari, Combatientes de Malvinas 3150, 1427 Buenos Aires, Argentina
e-mail: gabinemar@gmail.com
Resumen
La fibrosis quística se debe a la ausencia o defecto del canal transmembrana regulador de la fibrosis quística (CFTR), un canal de cloruro codificado en el gen cftr que juega un papel clave en la homeostasis del agua e iones. El CFTR es activado por el AMPc y se localiza en las membranas apicales y basolaterales de las vías aéreas, intestino y glándulas exocrinas. Una de sus funciones primarias en los pulmones es mantener la capa de líquido superficial a través de su función de canal y regular el canal epitelial de sodio sensible al amiloride (ENaC). Se han identificado más de 1900 mutaciones en el gen cftr. La enfermedad se caracteriza por secreciones viscosas en las glándulas exocrinas y por niveles elevados de cloruro de sodio en el sudor. En la fibrosis quística el CFTR no funciona y el ENaC está desregulado; el resultado es un aumento en la reabsorción de sodio y agua con la formación de un líquido viscoso. En las glándulas sudoríparas tanto el Na+ como el Cl- se retienen en el lumen causando una pérdida de electrolitos durante la sudoración y el NaCl se elimina al sudor. Así, los niveles elevados de NaCl son la base del test del sudor inducido por pilocarpina, un método de diagnóstico para la enfermedad. En esta revisión se discuten los movimientos de Cl- y Na+ en las glándulas sudoríparas y pulmón así como el papel del ENaC en la patogénesis de la enfermedad.
Palabras clave: Fibrosis quística; CFTR; ENaC; Movimientos iónicos; Citoesqueleto
Abstract
CFTR and ENaC functions in cystic fibrosis. Cystic fibrosis is caused by dysfunction or lack of the cystic fibrosis transmembrane conductance regulator (CFTR), a chloride channel that has a key role in maintaining ion and water homoeostasis in different tissues. CFTR is a cyclic AMP-activated Cl- channel found in the apical and basal plasma membrane of airway, intestinal, and exocrine epithelial cells. One of CFTR's primary roles in the lungs is to maintain homoeostasis of the airway surface liquid layer through its function as a chloride channel and its regulation of the epithelial sodium channel ENaC. More than 1900 CFTR mutations have been identified in the cftr gene. The disease is characterized by viscous secretions of the exocrine glands in multiple organs and elevated levels of sweat sodium chloride. In cystic fibrosis, salt and fluid absorption is prevented by the loss of CFTR and ENaC is not appropriately regulated, resulting in increased fluid and sodium resorption from the airways and formation of a contracted viscous surface liquid layer. In the sweat glands both Na+ and Cl- ions are retained in the lumen, causing significant loss of electrolytes during sweating. Thus, elevated sweat NaCl concentration is the basis of the classic pilocarpine-induced sweat test as a diagnostic feature of the disease. Here we discuss the ion movement of Cl- and Na+ ions in two tissues, sweat glands and in the air surface as well as the role of ENaC in the pathogenesis of cystic fibrosis.
Key words: Cystic fibrosis; CFTR; ENaC; Ionic movements; Cytoskeleton
Debido a la falla en el canal iónico CFTR (cystic fibrosis transmembrane conductance regulator) y a sus consecuencias en el canal de sodio sensible al amiloride (ENaC), se altera el transporte iónico en la fibrosis quística. Por un lado, la disminución en la entrada de Na+ y Cl- en los conductos de las glándulas sudoríparas aumenta el Na+ y Cl- en el sudor y es la base de la prueba diagnóstica más utilizada para la fibrosis quística. Por otro lado, la hiperabsorción del Na+ y la falta de secreción de HCO3- en el epitelio de las vías aéreas son responsables de la deshidratación del mucus y la consecuente cascada de fenómenos patológicos. Tratar de entender estos movimientos iónicos es el fundamento de este artículo.
La fibrosis quística es la enfermedad monogénica de carácter multisistémico más frecuente en la población mundial (1/3 000). Se hereda de manera autosómica recesiva. El defecto básico consiste en mutaciones en el gen que codifica la proteína CFTR, un canal aniónico expresado en células epiteliales y endoteliales del corazón, de la sangre y de placenta, entre otros. Además, el ENaC también juega un papel importante en la fisiopatología de la enfermedad.
El gen cftr se localiza en el cromosoma 7 (posición 7q31) y fue descubierto en 1989. Lo componen 300 000 pares de bases y 27 exones, está muy conservado entre las diferentes especies y por su tamaño ofrece un extenso blanco mutacional. Es así que se han detectado cerca de 1900 mutaciones en el gen que afectan el plegamiento, la localización o la actividad del canal. Dependiendo de la zona geográfica, el 25-90% de ellas consiste en la eliminación de un residuo de fenilalanina en la posición 508 dentro del dominio NBD1 (mutación ΔF508) que previene la inserción del canal en la membrana celular. En la Argentina, esta mutación se observa en el 59-66% de los afectados1-3. La segunda mutación más frecuente es la G542X, una mutación sin sentido que resulta en una proteína no funcional4. El elevado número de mutaciones dificulta la práctica de su identificación con métodos genéticos y es la razón por la cual todavía el "test del sudor" mediante iontoforesis de pilocarpina siga siendo un método de diagnóstico para la detección de la fibrosis quística5.
El transporte iónico defectuoso provoca una falla multiorgánica en órganos como pulmón, intestino y páncreas. En las vías aéreas, la pérdida del clearance mucociliar con el movimiento ciliar y la disminución de la capa de líquido de superficie favorece las infecciones recurrentes que llevan a una grave infección pulmonar responsable del 90% de la morbilidad y mortalidad. Hasta hace muy poco tiempo el tratamiento estuvo dirigido a las complicaciones de estos procesos patológicos, pero en los últimos años se han probado tratamientos en humanos con agentes que corrigen el funcionamiento del CFTR en enfermos con determinados tipos de mutaciones, última causa de la enfermedad (ver más adelante).
En forma experimental el CFTR se activa con drogas como la forskolina, un activador de la adenilato ciclasa, e inhibidores de la fosfodiesterasa, como el IBMX que aumenta los niveles de AMPc (Fig.1). El canal es inhibido por bloqueantes de canales de cloruro como el DPC y CFTRinh-1726 (Fig. 2).
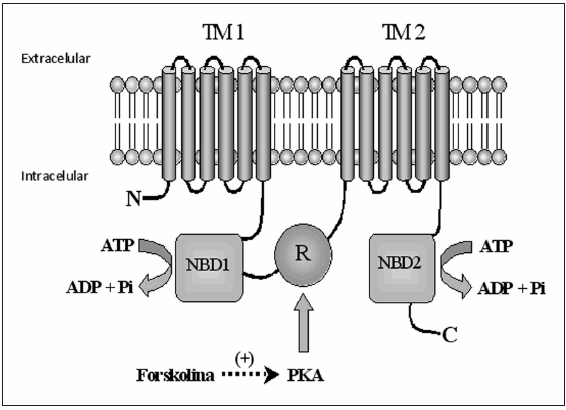
Fig. 1.- El CFTR es una glicoproteína con 1480 aminoácidos y un peso molecular de 180 KDa. Tiene 5 dominios, dos transmembranales (TM), cada uno con seis pasos de membrana alfa hélices unidos al dominio de unión a nucleótido (NBD) citoplasmático. El NBD1 está a su vez conectado a un sitio regulatorio R. El CFTR es activado por fosforilación de su dominio regulatorio intracelular R por la proteína quinasa dependiente de AMPc (PKA) que es activada por la forskolina37. Una vez que el canal es activado por la fosforilación, las aperturas y cierres son regulados por la hidrólisis del ATP en los dominios TM1 y TM2 y por el ATP intracelular que se une a los sitios NBD, siendo inactivado por fosfatasas.

Fig. 2.- Cambios en la conductancia (amplitud de corriente/amplitud del potencial) en un ovocito de Xenopus laevis inyectado 48 horas antes con el ARNm del CFTR. La célula es estimulada cada 30 segundos con pulsos despolarizantes de 60 mV de amplitud. La incubación del ovocito con forskolina aumenta la conductancia debido a la activación del CFTR y esto no se observa si se incuba a la célula con el bloqueante DPC. Por otro lado el DPC per se no tiene efecto sobre la conductancia básica de la célula. Las barras indican el tiempo de aplicación de las drogas y el valor de conductancia se relativiza respecto al valor basal.
Tanto en la síntesis, como en el plegamiento y degradación del canal intervienen numerosas moléculas chaperonas y todo este conjunto de proteínas, más de 200, se conoce como "CFTR interactoma"7, 8. El CFTR interacciona con diversas moléculas formando complejos macromoleculares anclados al citoesqueleto subcortical que permiten regular tanto la inserción del CFTR en la membrana, su nivel de expresión como su funcionamiento y degradación9. Un ejemplo está expuesto en la Fig. 3. El extremo C terminal del CFTR posee un dominio de unión de 4 aminoácidos (DTRL) para proteínas con dominios PDZ como NHERF (Na/H exchanger regulatory factor). Así, por ejemplo, la isoforma NHERF1, por medio de su extremo C terminal se une a EZRIN, una proteína estructural relacionada tanto a la membrana celular como a los filamentos de actina, estabilizando todo el complejo. Esto permite una compartamentalización celular y que el AMPc generado por PKA se mantenga en la zona del complejo. Por el contrario, en las mutaciones ΔF508 esta estructura está desintegrada, el AMPc está diseminado por todo el citoplasma y el canal mutado es degradado8.

Fig. 3.- Esquema de la interacción entre el CFTR y proteínas asociadas al citoesqueleto. El extremo C terminal del CFTR tiene un dominio de unión a los dominios PDZ (PDZBD) y de esta forma se une a la proteína NHERF1. A su vez NHERF1 por su extremo C terminal se une a EZRIN, una proteína estructural que se conecta tanto a la membrana celular como a los filamentos de actina que forman el citoesqueleto. Sólo se ha dibujado el TM 2, R, NBD2 y PDZBD del CFTR.
Como todas las proteínas, el CFTR se sintetiza y glicosila en el retículo endoplásmico transportado al aparato de Golgi, donde finaliza la glicosilación y es secretado hacia la membrana celular. Una vez en la membrana, el CFTR es reciclado en endosomas proximales y vueltos a la membrana celular y una pequeña fracción es transferida para su degradación a endosomas terminales, cuerpos multivesiculares y lisosomas10 (Fig. 4).

Fig. 4.- Esquema de la síntesis, maduración, inserción y degradación del CFTR. El CFTR se sintetiza en el retículo endotelial (RE) y completa su maduración y glicosilación en el sistema de Golgi para luego insertarse en la membrana plasmática donde cumple su función de canal iónico. Gran parte del CFTR es reciclado en vesículas y luego vuelto a la membrana plasmática mientras que otra fracción es degradada en los lisosomas.
Los potenciadores como el ivacaftor son compuestos de quinolinacarboxamida que mejoran la funcionalidad del canal insertado, mientras que los correctores brindan sus beneficios mediante el aumento del número de canales en la membrana por una disminución en su degradación, o facilitan su maduración o corrigen el defecto de los codones de finalización, como el ataluren que se utiliza en las mutaciones que tienen su fundamento en este mecanismo. El ivacaftor es un compuesto que aumenta la probabilidad de apertura del CFTR (estado abierto del canal) en los casos en que la fibrosis quística es causada por una mutación que provoca un bajo funcionamiento del canal (G551D). El ataluren es un aminoglicósido diseñado para promover la lectura en las mutaciones por codones de terminación como en la mutación G542X, restaurando la función del gen con resultados positivos en enfermos con fibrosis quística. El lumacaftor (VX-809) ha demostrado utilidad en cultivos promoviendo el tráfico hacia la membrana del CFTR con la mutación ΔF50811, 12 (Fig. 4). Existen así tratamientos novedosos para los dos tipos de mutaciones más frecuentes en Argentina.
Funciones del CFTR
1) En condiciones experimentales ha sido comprobado que tanto la proteína M2 del virus de la influenza, un canal protónico, así como la inflamación, hipoxia y factores oxidantes disminuyen la expresión y función del CFTR, sugiriendo un papel del CFTR en la fisiopatología de la influenza y en la enfermedad pulmonar obstructiva crónica13, 14.
2) Tanto el CFTR como el ENaC y otros canales iónicos participan en la migración celular, proceso crucial en la defensa inmunológica, la implantación y morfogénesis embrionaria, la reparación de heridas y la regeneración de los tejidos. Es un fenómeno complejo que requiere de la integración de señales mecánicas y químicas que reprimen la expresión de moléculas de adhesión y producen cambios en el volumen y en el citoesqueleto celular. La regulación aberrante de los canales iónicos es un mecanismo que subyace en la invasión de los tumores malignos15, 16.
3) Además de su función como canal iónico de cloruro, el CFTR puede transportar HCO3- y su defecto es muy importante en la fisiopatología de la fibrosis quística (ver más adelante)17, 18.
4) El CFTR puede ser activado por estímulos mecánicos y se ha caracterizado así como un canal mecanosensible, particularidad importante en los cambios de volumen celular en los que participa el CFTR19.
5) Existen numerosas pruebas acerca de que el CFTR regula la actividad de otros canales iónicos como canales de cloruro de rectificación saliente (ORCC), el ENaC y las conexinas20-22. Ejemplo de ello se observa en la Fig. 5 con la disminución en la conductancia a través de las uniones gap por el CFTR activado por forskolina en ovocitos que expresan también la conexina 45, proteína formadora de las uniones gap. La actividad del CFTR altera la respuesta control. Aunque los detalles de esta regulación no se conocen, los mecanismos podrían involucrar cambios en la concentración de iones por la actividad de un canal que a su vez afectaría a otro canal o en el número de canales expresados. Una interacción física directa entre CFTR y ENaC se apoya en los resultados obtenidos en experimentos de electrofisiología de canales aislados, la coinmunoprecipitación de las dos proteínas y por la presencia del fenómeno FRET (transmisión de energía de resonancia de Förster) que requiere de una proximidad de las proteínas de 1-10-nm23, 24 (Fig. 5).

Fig. 5.- Interacción entre CFTR y la conexina 45 en pares de ovocitos acoplados para que se formen las uniones gap y que expresan 1) la conexina 45 o 2) el CFTR más la conexina 45. La estimulación del CFTR con forskolina disminuye el cociente del valor estacionario de la conductancia (Gjss) respecto a la del pico de la misma (GJmax) cuando uno de los ovocitos es estimulado (transjunctional voltage, Vj) con pulsos positivos y negativos de 0 a 80 mV. (Modificado de Kotsias y Peracchia, 2005)22.
En 1986 se publicó un artículo25 en el que se demostraba en la fibrosis quística una hiperabsorción de Na+ en el epitelio de las vías aéreas, sugiriéndose la participación del canal ENaC en el desarrollo de la enfermedad. A partir de ese momento se publicaron nuevas evidencias y contradicciones sobre la relación del ENaC con el CFTR y del papel regulador del CFTR sobre otros canales, que incluyeron modelos transgénicos de ratones con falta de CFTR y sobreexpresión de una de las subunidades del ENaC26, 27. Como se comentó más arriba, faltan muchos detalles sobre esta interacción, en particular sobre sus efectos positivos y negativos dependiendo del tejido estudiado. El lector es referido a dos revisiones completas sobre el tema28, 29. Aquí nos detendremos en uno de esos mecanismos que podrían explicar los dos puntos mencionados en la primera parte de este trabajo como el aumento en el Na+ y Cl- en el sudor de los enfermos con fibrosis quística y la hiperabsorción de Na+ en las vías aéreas enfermas.
En líneas generales, la absorción de los iones y agua depende de los efectos del transporte activo de Na+ en las membranas basolaterales. El Cl- y el Na+ se transportan al interior celular por varios mecanismos, incluyendo el CFTR y el canal ENaC en respuesta a cambios en la fuerza impulsora creada por la NaK-ATPasa. El movimiento de iones tiene por resultado el pasaje de agua por vía paracelular y transcelular30, 31.
En la Fig. 6 se muestra un diagrama de esta interacción en las glándulas sudoríparas. En condiciones normales tanto el Cl- como el Na+ se reabsorben siguiendo el gradiente electroquímico. Cuando la función del CFTR es defectuosa o no está presente como en la fibrosis quística, el Cl- del lumen en los conductos no se reabsorbe y este exceso de cargas negativas en el exterior celular despolariza la membrana apical dificultando la reabsorción de Na+ por la atracción entre cargas opuestas. Como consecuencia tanto el Cl- como el Na+ se acumulan en la luz de los conductos y de allí la mayor concentración de ambos en el sudor de los enfermos, y la base del test del sudor.

Fig. 6.- Principales formas de transporte de Na+ y Cl- en glándulas sudoríparas y vía aérea. El Na+ y el Cl- se movilizan por los canales ENaC y CFTR en la membrana apical (MA) y basolateral (MBL). El transporte activo (TA) de la NaK-ATPasa crea el gradiente para la movilización de los iones y el cotransportador Na-K-2Cl es una de las fuentes de entrada que permite que la concentración de Cl- esté por encima de su potencial electroquímico. Los inhibidores de las proteasas del líquido de superficie y la proteína SPLUNC1 tienen un efecto negativo sobre el ENaC, disminuyendo su pasaje al interior celular y el cambio en el potencial de membrana influye sobre el pasaje de Cl- por el CFTR. La interacción positiva que se observa en la fibrosis quística entre el defecto del CFTR y el ENaC está simbolizada por el signo positivo. Ver texto para más detalles.
En las vías aéreas la concentración intracelular de Cl- es más elevada que la determinada por el potencial electroquímico debido a su entrada por un cotransportador de la membrana basolateral que utiliza los gradientes de Na+, K+ y Cl- generados por la NaK- ATPasa. Además, las células del epitelio respiratorio poseen una mayor permeabilidad al agua que en las glándulas sudoríparas y pueden alternar entre la secreción y la reabsorción del Cl- dependiendo del gradiente electroquímico; para esto se utilizan canales CFTR ubicados tanto en la membrana apical como en la basolateral y una regulación del ENaC por factores extracelulares muy importantes:
- En primer lugar la presencia de una proteína secretada, SPLUNC1 (short palate lung and nasal epithelial clone 1) que inhibe la función del ENaC cuando el pH extracelular es cercano a la neutralidad32. En la fibrosis quística el transporte de HCO3- está afectado y el pH extracelular es más ácido y esta acidez es responsable de que SPLUNC1 no bloquee el ENaC con la consiguiente hiperabsorción de Na+ y consiguiente deshidratación.
- En segundo lugar, la presencia de inhibidores de proteasas en el líquido superficial de la vía aérea debido a que las proteasas activan al ENaC33. Una de las proteasas es la elastasa, con una mayor actividad durante los estados inflamatorios causando un incremento en la absorción de Na+ al activarse los canales ENaC. Debido a este fenómeno se deshidratan las vías aéreas con disminución de la actividad ciliar34. Cuando la capa de líquido disminuye por algún factor, por ejemplo en la deshidratación, aumenta la concentración de los inhibidores de proteasas y el transporte de Na+ disminuye. Este exceso de cargas permite que el Cl- se secrete tratando de recomponer el líquido.
En la fibrosis quística por la falla en el CFTR estas regulaciones en el transporte de Cl- no ocurren con una hiperabsorción de Na+ y pasaje de agua con afectación en la composición y cantidad del líquido de la superficie aérea32. La función del CFTR y su consecuencia en el transporte de Na+ puede ser estudiada in vivo en los enfermos midiendo la diferencia del potencial nasal, un método de diagnóstico clínico35.
Es importante mencionar que existen todavía preguntas y contradicciones sin respuestas ni explicaciones. Por ejemplo, atendiendo al papel clave de la hiperabsorción de Na+ en la fisiopatogenia de la enfermedad pulmonar, han fracasado los intentos terapéuticos utilizando bloqueantes del ENaC como el amiloride36. Por otro lado, un modelo de ratón con la mutación más frecuente en humanos, la ΔF508, produce una enfermedad intestinal pero no pulmonar26, mientras que en el modelo de ratón transgénico con sobreexpresión del ENaC la enfermedad pulmonar se desarrolla aun cuando el CFTR endógeno está presente27.
A partir de nuestros estudios in vitro tenemos suficientes evidencias que el ENaC y el CFTR expresados en la placenta y en la línea celular BeWo derivada del trofoblasto, participan en la migración de las células placentarias15, 16. El ENaC es un canal mecanorreceptor, regulado principalmente por la aldosterona, se coexpresa con el CFTR, e interactúa y está en relación directa con proteínas del citoesqueleto. Su desregulación o menor expresión, como hemos demostrado en placentas preeclámpticas38 al igual que con el CFTR39 podrían ser importantes para explicar uno de los mecanismos alterados en la preeclampsia, como es la menor migración e invasión del trofoblasto.
Cuando el gen responsable se aisló en 198940 se pensó que el tratamiento de la enfermedad estaba cercano. Ahora sabemos, a un cuarto de siglo de este hallazgo, que la enfermedad ha contribuido más al conocimiento científico que la ciencia a la cura de la enfermedad. Más de 1900 mutaciones sobre el mismo gen, la compleja fisiopatogenia por la participación del canal ENaC, la diferencia entre los modelos animales con la enfermedad y las complejas regulaciones e interacciones entre los canales iónicos son algunos de los factores responsables para el retardo experimentado en el tratamiento dirigido a la base de la enfermedad y no a sus consecuencias.
Conflictos de intereses. Este trabajo fue financiado con subsidios del Ministerio de Ciencia Tecnología e Innovación Productiva, Universidad de Buenos Aires, Fundación A. Roemmers y Consejo Nacional de Investigaciones Científicas y Técnicas.
1. Saleh MC, Botelli A, Melano de Botelli M, Rezzonico CA, Argaraña CE. Cystic fibrosis: frequency of delta f508 and g542x mutations in Cordoba, Argentina. Medicina (B Aires) 1996; 56: 14-6. [ Links ]
2. Pérez MM, Luna MC, Pivetta OH, Keyeux G. CFTR gene analysis in Latin American CF patients: heterogeneous origin and distribution of mutations across the continent. J Cyst Fibros 2007; 6: 194-208. [ Links ]
3. Oller de Ramírez AM, Ghio A, Melano de Botelli M, Dodelson de Kremer R. Fibrosis quística: diagnóstico molecular en 93 pacientes argentinos y detección familiar de portadores. Impacto asistencial y proyección a nuevos avances terapéuticos. Arch Argent Pediatr 2008; 106: 310-9 [ Links ]
4. Levy EM, Granados P, Rawe V et al. Mutations in CFTR gene and clinical correlation in Argentine patients with congenital bilateral absence of the vas deferens. Medicina (B Aires) 2004; 64: 213-8. [ Links ]
5. Mattar AC, Leone C, Rodrigues JC, Adde FV. Sweat conductivity: An accurate diagnostic test for cystic fibrosis? J Cyst Fibros 2014 Jan 30. pii: S1569-1993(14)00004-6. doi: 10.1016/j.jcf.2014.01.002. [Epub ahead of print]. [ Links ]
6. Ma T, Thiagarajah JR, Yang H et al. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest 2002; 110: 1651-8. [ Links ]
7. Chanoux RA, Rubenstein RC. Molecular chaperones as targets to circumvent the CFTR defect in cystic fibrosis. Front Pharmacol 2012; 3:137. doi: 10.3389/fphar.2012.00137. eCollection 2012. [ Links ]
8. Monterisi S, Casavola V, Zaccolo M. Local modulation of cystic fibrosis conductance regulator: cytoskeleton and compartmentalized cAMP signalling. Br J Pharmacol 2013; 169: 1-9. [ Links ]
9. Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol 2006; 7: 426-36. [ Links ]
10. Cholon DM, O'Neal WK, Randell SH, Riordan JR, Gentzsch M. Modulation of endocytic trafficking and apical stability of CFTR in primary human airway epithelial cultures. Am J Physiol Lung Cell Mol Physiol 2010; 298: L304-14. [ Links ]
11. Peltz SW, Morsy M, Welch EM, Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med 2013; 64: 407-25. [ Links ]
12. Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med 2013; 1: 158-63. [ Links ]
13. Londino JD, Lazrak A, Jurkuvenaite A, Collawn JF, Noah JW, Matalon S. Influenza matrix protein 2 alters CFTR expression and function through its ion channel activity. Am J Physiol Lung Cell Mol Physiol 2013; 304: L582-92. [ Links ]
14. Rab A, Rowe SM, Raju SV, Bebok Z, Matalon S, Collawn JF. Cigarette smoke and CFTR: implications in the pathogenesis of COPD. Am J Physiol Lung Cell Mol Physiol 2013; 305: L530-41. [ Links ]
15. Marino GI, Kotsias BA. Cystic fibrosis transmembrane regulator (CFTR) in human trophoblast BeWo cells and its relation to cell migration. Placenta 2014; 35: 92-8. [ Links ]
16. Marino GI, Kotsias BA. The epithelial sodium channel and the cell migration. Physiological Mini Reviews 2012; 6: 13-22. [ Links ]
17. Tang L, Fatehi M, Linsdell P. Mechanism of direct bicarbonate transport by the CFTR anion channel. J Cyst Fibros 2009; 8: 115-21. [ Links ]
18. Shcheynikov N, Kim KH, Kim KM et al. Dynamic control of cystic fibrosis transmembrane conductance regulator Cl(-)/HCO3(-) selectivity by external Cl(-). J Biol Chem 2004; 279: 21857-65. [ Links ]
19. Zhang WK, Wang D, Duan Y, Loy MM, Chan HC, Huang P. Mechanosensitive gating of CFTR. Nature Cell Biol 2010; 12: 507-12. [ Links ]
20. Reddy MM, Quinton PM. ENaC activity requires CFTR channel function independently of phosphorylation in sweat duct. J Membr Biol 2005; 207: 23-33. [ Links ]
21. Kotsias BA, Salim M, Peracchia LL, Peracchia C. Interplay between cystic fibrosis transmembrane regulator and gap junction channels made of connexins 45, 40, 32 and 50 expressed in oocytes. J Membr Biol 2006;214: 1-8. [ Links ]
22. Kotsias BA, Peracchia C. Functional interaction between CFTR and Cx45 gap junction channels expressed in oocytes. J Membr Biol 2005; 203: 143-50. [ Links ]
23. Berdiev BK, Qadri YJ, Benos DJ. Assessment of the CFTR and ENaC association. Mol Biosyst 2009; 5: 123-7. [ Links ]
24. Qadri YJ, Cormet-Boyaka E, Benos DJ, Berdiev BK. CFTR regulation of epithelial sodium channel. Methods Mol Biol 2011; 742: 35-50. [ Links ]
25. Boucher RC, Stutts MJ, Knowles MR, Cantley L, Gatzy JT. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J Clin Invest 1986; 78: 1245-52. [ Links ]
26. Zeiher BG, Eichwald E, Zabner J et al. A mouse model for the delta F508 allele of cystic fibrosis. J Clin Invest 1995; 96: 2051-64. [ Links ]
27. Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na absorption produces cystic fibrosis-like lung disease in mice. Nat Med 2004; 10: 487-93. [ Links ]
28. Collawn JF, Lazrak A, Bebok Z, Matalon S. The CFTR and ENaC debate: how important is ENaC in CF lung disease? Am J Physiol Lung Cell Mol Physiol 2012; 302: L1141-6. [ Links ]
29. Hobbs CA, Da Tan C, Tarran R. Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? J Physiol 2013; 591: 4377-87. [ Links ]
30. Reddy MM, Stutts MJ. Status of fluid and electrolyte absorption in cystic fibrosis. Cold Spring Harb Perspect Med 2013; 3:a009555.doi: 10.1101/cshperspect.a009555. [ Links ]
31. Frizzell RA, Hanrahan JW. Physiology of epithelial chloride and fluid secretion. Cold Spring Harb Perspect Med 2012; 2:a009563. doi: 10.1101/cshperspect.a009563. [ Links ]
32. Garland AL, Walton WG, Coakley RD et al. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc Natl Acad Sci U S A 2013; 110: 15973-8. [ Links ]
33. Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, Kleyman TR. Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J Biol Chem 2008; 283: 36586-91. [ Links ]
34. Prulière-Escabasse V, Clerici C, Vuagniaux G, Coste A, Escudier E, Planès C. Effect of neutrophil elastase and its inhibitor EPI-hNE4 on transepithelial sodium transport across normal and cystic fibrosis human nasal epithelial cells. Respiratory Res 2010; 11: 141. doi: 10.1186/1465-9921-11-141. [ Links ]
35. Rowe SM, Clancy JP, Wilschanski M. Nasal potential difference measurements to assess CFTR ion channel activity. Methods Mol Biol 2011; 741: 69-86. [ Links ]
36. Pons G, Marchand MC, d'Athis P et al. French multicenter randomized double-blind placebo-controlled trial on nebulized amiloride in cystic fibrosis patients. The Amiloride-AFLM Collaborative Study Group. Pediatr Pulmonol 2000; 30: 25-31. [ Links ]
37. King JD Jr, Lee J, Riemen CE et al. Role of binding and nucleoside diphosphate kinase A in the regulation of the cystic fibrosis transmembrane conductance regulator by AMP-activated protein kinase. J Biol Chem 2012; 287: 33389-400. [ Links ]
38. Marino GI, Kotsias BA. Expression of the epithelial sodium channel sensitive to amiloride (ENaC) in normal and preeclamptic human placenta. Placenta 2013; 34: 197-200. [ Links ]
39. Castro-Parodi M1, Levi L, Dietrich V, Zotta E, Damiano AE. CFTR may modulate AQP9 functionality in preeclamptic placentas. Placenta 2009; 30: 642-8. [ Links ]
40. Riordan JR, Rommens JM, Kerem B et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066-73. [ Links ]
Recibido: 29-I-2014
Aceptado: 5-III-2014

