










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680
Medicina (B. Aires) vol.74 no.5 Ciudad Autónoma de Buenos Aires Oct. 2014
ARTÍCULO ESPECIAL
Inmunopatología de la esclerosis múltiple
Francisco J. Quintana1, Soledad Pérez-Sánchez2 y Mauricio F. Farez3
1Center for Neurologic Diseases, Department of Neurology, Brigham and Women's Hospital, Harvard Medical School, Boston, USA,
2Unidad de Esclerosis Múltiple, Hospital Universitario Virgen Macarena, Sevilla, España,
3Instituto de Investigaciones Neurológicas Dr. Raúl Carrea, FLENI, Buenos Aires, Argentina
Dirección postal: Francisco J. Quintana, Center for Neurologic Diseases, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 714, Boston, MA 02115, USA
Fax: (001) 617 525 5305 e-mail: fquintana@rics.bwh.harvard.edu
Resumen
La esclerosis múltiple es una enfermedad inflamatoria desmielinizante que afecta el sistema nervioso central y que es considerada una de las principales causas de discapacidad en jóvenes adultos. Las causas de la esclerosis múltiple son aún desconocidas, aunque se cree que una combinación de factores genéticos y ambientales resulta en una respuesta autoinmune que promueve la degeneración neuronal/axonal. En esta revisión se analiza la asociación entre la respuesta inmune y la neurodegeneración en la esclerosis múltiple.
Palabras clave: Esclerosis múltiple; Patogénesis.
Abstract
Immunopathology of multiple sclerosis. Multiple sclerosis is an inflammatory demyelinating disease affecting the central nervous system and considered one of the leading causes of disability in young adults. The precise cause of multiple sclerosis is unknown, although the current evidence points towards a combination of genetic and environmental factors leading to an autoimmune response that promotes neuronal degeneration. In this review, we will describe the association between the immune response and neurodegeneration in multiple sclerosis.
Key words: Multiple sclerosis; Pathogenesis.
La esclerosis múltiple (EM) es una enfermedad inflamatoria desmielinizante que afecta el sistema nervioso central (SNC) y es una de las principales causas de discapacidad en adultos jóvenes. En el 85% de los casos, la enfermedad se manifiesta inicialmente con un curso de brotes seguidos de remisiones totales o parciales, etapa denominada esclerosis múltiple con recaídas y remisiones (EMRR), y es la fase de la enfermedad que mejor respuesta clínica presenta a las intervenciones terapéuticas disponibles. Eventualmente, el curso clínico de la enfermedad evoluciona, mostrando un deterioro neurológico progresivo independiente de las recaídas y remisiones con una respuesta limitada a los tratamientos, y se denomina esclerosis múltiple secundaria progresiva (EMSP). Finalmente, en un grupo reducido de pacientes la enfermedad presenta desde su diagnóstico un curso de deterioro progresivo de la función neurológica, referido como esclerosis múltiple progresiva primaria (EMPP).
La EM se caracteriza por la presencia en el SNC de lesiones (placas) inflamatorias desmielinizadas que se caracterizan por la disrupción de la barrera hematoencefálica, inflamación, desmielinización, pérdida de oligodendrocitos, gliosis reactiva y degeneración neuronal/axonal1-3, siendo esta última la causa más importante de discapacidad neurológica en la EM4. En esta revisión analizaremos la asociación entre la respuesta inmune y la neurodegeneración en la EM, enfocándonos en los mecanismos efectores de la respuesta inmune.
Mecanismos inmunopatológicos en la esclerosis múltiple
Células T CD4+
Las terapias que impiden la entrada de células T al SNC, como el natalizumab5-7 o que inhiben la salida de células T de los nódulos linfáticos (fingolimod)8-11 tienen fuertes efectos terapéuticos en la EMRR. La administración del alemtuzumab, un anticuerpo anti-CD52 que elimina células T CD4+ y CD8+ de la circulación, resulta en una significativa disminución en los brotes y las nuevas lesiones8-11. Estas observaciones sugieren que las células T juegan un rol importante en esta etapa de la enfermedad.
Entre las más importantes células T CD4+ efectoras involucradas en la patología de la EM se encuentran las células Th1 y Th17. Las células Th1 se diferencian en respuesta a la activación en presencia de la interleuquina 12 (IL-12), y se caracterizan por la expresión del factor de transcripción Tbet, el cual controla un programa de expresión génica que resulta en la producción interferón gamma (IFNγ) y otras moléculas efectoras12, 13 (Fig. 1). Las células Th17 se diferencian en respuesta a la activación en presencia del factor de crecimiento transformante β1 (TGFβ1), IL-6 o IL-21 e IL-23, y se caracterizan por la expresión del factor de transcripción RORγt, que controla un programa de expresión génica que resulta en la expresión de IL-17 y otras moléculas efectoras14-16.

Fig. 1. Vías inmunológicas en la inflamación y neurodegeneración de la EM. La activación de células T CD4+ por las células presentadoras de antígenos y en presencia de diversas citoquinas, lleva a su diferenciación a varios subtipos, entre ellos las células Th1 y Th17, que a través de la secreción de citoquinas, estimulación de microglia y astrocitos, llevan a la formación de la placa desmielinizada. A su vez, la activación de microglia y astrocitos, junto con la activación de las células T CD8+ a través de varios mecanismos, llevan a un mayor daño axonal y eventualmente contribuyen con la neurodegeneración observada en la EM.
TGFB-1: transforming growth factor beta 1; TNF: tumor necrosis factor; IFN: interferón; MCH: Complejo mayor de histocompatibilidad.
Las células Th1 y Th17 contribuyen por distintos mecanismos a la patología de la EM. A pesar de que otras células del sistema inmune pueden producirlas, IFNγ e IL-17, citoquinas clásicamente usadas para definir a las células Th1 y Th17 respectivamente, tienen efectos directos en la patología de la EM. Panitch y colaboradores administraron IFNγ a 18 pacientes con EMRR y observaron la inducción de brotes en 7 de esos pacientes17, 18, sugiriendo que el IFNγ contribuye a la patología de la EM. Por otra parte, la administración de secukinumab (en fase IIa, un anticuerpo que neutraliza la IL-17), reduce significativamente el número de lesiones en el SNC y muestra una tendencia a disminuir el número de brotes durante 6 meses19, 20.
Cabe destacar que las células Th1 y Th17 también promueven la activación de microglia, macrófagos, astrocitos y linfocitos B a través de la producción de citoquinas y factores de crecimiento, activando consecuentemente mecanismos adicionales neurodegenerativos. Por ejemplo, las células Th1 y Th17 producen GM-CSF, el cual activa funciones neurodegenerativas en microglia21-23. Finalmente la proteína podoplanina, producida por las células Th17, promueve la formación de nódulos linfáticos terciarios en el SNC en los cuales se establecen y diferencian células productoras de anticuerpos24, 25.
Células T CD8+
Las células CD8+ son 3-10 veces más abundantes que las CD4+ en placas crónicamente inflamadas en el CNS de enfermos con EM26-29. El daño axonal correlaciona más fuertemente con el número de células T CD8+ y microglia/macrófagos que con las CD4+30, 31. De hecho, las células T CD8+ se localizan y expanden clonalmente tanto en las lesiones perivasculares del CNS en MS como en el parénquima, mientras que las células T CD4+ están mayoritariamente restrictas a las regiones perivasculares26, 32. Además, las células T CD8+ inducen muerte neuronal en cultivo33, 34. Estas observaciones sugieren que las células CD8+ también participan en la patología de la EM29.
Las células T CD8+ interaccionan con células que expresan el complejo mayor de histocompatibilidad de clase I (MHCI), el cual es expresado por todas las células nucleadas32, formando una sinapsis inmunológica estabilizada por las moléculas de adhesión LFA-1 e ICAM-129. Diversos mecanismos están involucrados en la destrucción de neuronas por células T CD8+. La citotoxicidad por células T CD8+ es mediada in vivo mayoritariamente por dos mecanismos: 1) La secreción de gránulos líticos que contienen perforina y granzimas, las cuales pueden disparar la ruptura de la membrana celular y/o apoptosis. 2) La interacción de FasL con Fas expresado en neuronas34, 35. Diferencias en la intensidad de la interacción MHC/TCR favorecen el uso de un mecanismo específico de citotoxicidad29. Sin embargo, es probable que in vivo todos estos mecanismos contribuyan a los efectos patogénicos de las células T CD8+ en neuronas.
En el contexto de la neuroinflamación, es importante considerar que las células T CD8+ producen también grandes cantidades de TNFα e IFNγ. El TNFα altera directamente la estructura y funcionalidad de la membrana neuronal, interfiriendo con la funcionalidad de las neuronas36, 37 e induciendo su apoptosis38, 39. El IFNγ modula la actividad del receptor AMPA GluR1, incrementando la muerte neuronal por excitotoxicidad40. Finalmente, células T CD8+ que producen IL-17 también han sido identificados en el SNC de pacientes con EM, sugiriendo que la IL-17 producida por estas poblaciones celulares también participa en la patogenia de la enfermedad y a los efectos terapéuticos del secukinumab.
Células B
Los resultados clínicos positivos observados con el uso de rituximab para el tratamiento de la EM, un anticuerpo monoclonal anti-CD20 que elimina los linfocitos B circulantes, sugieren que las células B juegan un importante rol en la patología de la enfermedad41. Curiosamente, el tratamiento con rituximab reduce el número de células B, pero no las bandas oligoclonales o la concentración de anticuerpos en el SNC, lo que sugiere que los efectos beneficiosos del tratamiento están asociados a la depleción de linfocitos B y no a la modificación de los niveles de autoanticuerpos. Como resultado de estas observaciones, se considera que el principal aporte de las células B a la patología de la EM es a través de la producción de citoquinas pro-inflamatorias como la linfotoxina y el TNFα, y su capacidad de actuar como células presentadoras de antígeno para activar células T. Esta hipótesis es respaldada por la disminución en la frecuencia de células patogénicas Th1 y Th17 observada en enfermos tratados con rituximab42, 43.
A pesar que los efectos terapéuticos del rituximab no están asociados a la eliminación de anticuerpos, autoanticuerpos reactivos con el SNC participan en la patología de la EM en determinadas subpoblaciones de enfermos. Anticuerpos dirigidos contra epítopes conformacionales de proteínas de mielina son detectados en pacientes con EM, inclusive en etapas muy tempranas de la enfermedad25, 44-47. La patogenicidad de estos anticuerpos ha sido demostrada en diversos sistemas experimentales48, 49.
Las observaciones antes mencionadas sugieren efectos patogénicos directos de autoanticuerpos en la progresión de la enfermedad. Sin embargo, además de su potencial contribución a la patología de la EM, los anticuerpos ofrecen una ventana para estudiar la respuesta inmune y su respuesta al tratamiento: nuestro grupo ha demostrado que el estudio de la respuesta de anticuerpos usando microchips de antígenos permite la estratificación de los pacientes con EM, el análisis de la respuesta inmune local en el SNC y el monitoreo de la respuesta al tratamiento44.
Microglia y macrófagos inflamatorios
La microglia, los macrófagos residentes del SNC, constituyen aproximadamente el 10% de las células del SNC50. Las células de la microglia se encuentran constantemente abocadas a la remoción de desechos celulares y a la detección de patógenos en el SNC. Al activarse en respuesta a lesiones, inflamación o infecciones, la microglia cambia su aspecto morfológico tomando un aspecto ameboide, y aumenta la expresión de marcadores de superficie típicamente asociados a macrófagos como F4/80 y Mac-1. Sin embargo, el estímulo específico involucrado (citoquinas, agonistas de receptores tipo Toll) determina el fenotipo funcional que toma la microglia luego de su activación: este fenotipo puede ser pro-inflamatorio (fenotipo M1) o anti-inflamatorio y asociado al remodelado de tejidos y cicatrización (fenotipo M2)51,52. Estos fenotipos están asociados a programas transcripcionales específicos53 pero, sin embargo, representan extremos de espectro de posibles fenotipos interconvertibles in vivo.
En los estadios tempranos de EM, grupos de microglia activada y macrófagos periféricos reclutados al SNC pueden identificarse en las lesiones co-localizados con daño axonal y neuronal54, 55. La microglia y los macrófagos son activados por citoquinas producidas por las células T y también por productos de la degradación de mielina56, 57. La activación de células de microglia y macrófagos resulta en la producción de citoquinas, chemoquinas y metabolitos que regulan directa e indirectamente la neurodegeneración en la EM50, 51, 58, 59. La chemoquina CCL-2 producida por microglia activada, por ejemplo, afecta la integridad de la barrera hematoencefálica y atrae macrófagos periféricos al SNC. A su vez, ya reclutados al SNC, los macrófagos pueden adquirir también un fenotipo pro-inflamatorio (M1) que promueve la neurodegeneración. La microglia y los macrófagos M1 producen las citoquinas IL-12 e IL-23, las cuales contribuyen a la diferenciación de células Th1 y Th17 respectivamente. Además, las células de la microglia y los macrófagos expresan moléculas MHCI y MHCII junto con moléculas co-estimuladoras CD40, CD80, CD86, lo que les permite reactivar células T en el SNC, promoviendo la diferenciación de células patogénicas Th1 y Th17.
La microglia y macrófagos producen también moléculas con directa actividad neurotóxica. El TNFα induce apoptosis en neuronas y también actúa en forma autócrina para promover la secreción de glutamato, incrementando la muerte neuronal causada por excitotoxicidad60. LaIL-1β también tiene actividades neurotóxicas, e induce la producción de óxido nítrico (ON), que junto con las especies reactivas de oxigeno (ERO), favorece la neurotoxicidad.
Astrocitos
Los astrocitos constituyen el más abundante y diverso tipo de células de la glía en el SNC, a cargo de importantes funciones metabólicas e inmunológicas61. Los astrocitos perivasculares presentan un daño significativo en lesiones activas en EM; este daño sugiere que las disfunciones en la barrera hematoencefálica que caracterizan la enfermedad están asociadas a defectos en la funcionalidad de astrocitos62. Durante el curso de la EM, distintos estímulos como citoquinas y productos de degradación de la mielina producen la activación de los astrocitos, resultando en la producción de citoquinas y chemoquinas que promueven la respuesta inflamatoria en el SNC57, 61-63.
Los astrocitos son una fuente importante de la chemoquina CCL-2, que recluta macrófagos inflamatorios al SNC, y también de TNFα, que promueve la apoptosis en neuronas57, 61-63. Los astrocitos producen cantidades biológicamente significativas de ON, ERO, glutamato y ATP en las lesiones en EM62, que al interferir con la actividad mitocondrial en neuronas promueven la pérdida de axones y neuronas. La secreción de ATP tiene también importantes efectos para la regulación de la respuesta inmune, activando respuestas pro-inflamatorias en distintos tipos celulares como microglia y células dendríticas64, 65 y disparando además efectos neurotóxicos directos66. La secreción de glutamato, acompañada de una reducida capacidad de limitar los niveles extracelulares de glutamato observada en astrocitos en EM, resulta en un incremento en la muerte neuronal inducida por exitotoxicidad57, 67.
Finalmente, los astrocitos regulan la actividad de otras células involucradas en la inmunopatología de la EM, influenciando la actividad de oligodendrocitos, células T, microglia y macrófagos, células B, células dendríticas, células NK y células T γδ68.
Oligodendrocitos
Los oligodendrocitos (OLs) son células de la glía que controlan la producción y mantenimiento de la mielina en el SNC69. Los OLs se diferencian a partir de las células precursoras de OLs (OPC) durante las primeras etapas del desarrollo, aunque las OPCs mantienen su capacidad de diferenciación en OLs en el SNC adulto69 (Fig. 2). De hecho, las OPC tienen la capacidad de proliferar y diferenciarse en respuesta a distintos estímulos tóxicos, traumáticos o inflamatorios, pero esta capacidad se pierde gradualmente durante el envejecimiento70. Sin embargo, diversas vías de señalización regulan positiva y negativamente la diferenciación de OLs69. La regulación de dichas vías es considerada una potencial estrategia terapéutica para promover la remielinización en EM y detener y revertir la disfunción neurológica.
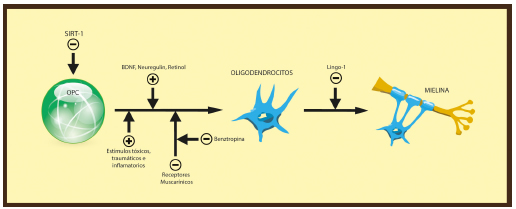
Fig. 2. Mecanismos de regulación de la remielinización en la EM. Vías de señalización que regulan positiva (BDNF, neuregulin, retinol, diversos estímulos tóxicos y traumáticos) y negativamente (sirtuina-1, receptores muscarínicos) la diferenciación de OPCs en OLs. LINGO-1 inhibe la diferenciación de OLs y la mielinización a través de su interacción con receptores NgR1.
SIRT-1: sirtuina-1; OPCs; OLs: Células precursoras de oligodendrocitos; oligodendrocitos BDNF: factor neurotrófico derivado del cerebro; Lingo-1(o LINGO-1): Leucine rich repeat and Ig domain containing 1.
Nota: Las figuras pueden observarse en color en www.medicinabuenosaires.com
LINGO-1 es una glicoproteína de superficie expresada en neuronas y OLs que inhibe la diferenciación de OLs y la mielinización a través de su interacción con los receptores NgR1. Consecuentemente, nuevas terapias están siendo desarrolladas para promover la remielinización en EM basadas en el bloqueo de las vías de señalización activadas por LINGO-1. Un anticuerpo monoclonal neutralizante contra LINGO-1 (BIIB033) se encuentra actualmente en ensayo clínico para el tratamiento de EMRR71. La sirtuina-1 también tiene un efecto inhibitorio en la remielinización, sugiriendo que terapias dirigidas a bloquear sirtuina-1 pueden promover la remielinización en EM.
Entre las vías activadoras naturales de la remielinización se encuentran el retinol y su receptores gamma72. Factores de crecimiento como la neuregulin o BDNF también pueden promover la mielinización73. El conocimiento de todas las vías y moléculas implicadas en el complejo proceso de remielinización son esenciales para desarrollar dianas terapéuticas y restablecer la funcionalidad de las lesiones desmielinizadas.
La neurodegeneraci ón y la inflamación están intrínsecamente ligadas al curso de la EM. La activación desmedida del sistema inmune promueve la neurodegeneración, y el daño neuronal resulta en la liberación de nuevos epítopes antigénicos y moléculas pro-inflamatorias que aumentan y perpetúan la respuesta inmune en el SNC. Estas observaciones sugieren que el tratamiento efectivo de los pacientes con EM requiere el control tanto del componente neurodegenerativo como del componente inflamatorio de la enfermedad. Las intervenciones terapéuticas disponibles en la actualidad modulan principalmente los aspectos inmunológicos de la enfermedad, y dentro de ellos solo aquellos relacionados con la respuesta inmune adaptativa (células B y T), generalmente en una forma antígeno inespecífica. El reto para nuevas terapias de la EM es la inducción de tolerancia inmune antígeno-específica, por ejemplo a través del uso de protocolos de tolerización con péptidos74, 75, vacunas de ADN76-82 o nanopartículas83, 84. Además, las futuras terapias para la EM deben estar dirigidas también a controlar los componentes innatos del sistema inmune (microglía, macrófagos, astrocitos)57, 63, 67 y a promover la remielinización. Un enfoque terapéutico combinado, que controle los componentes inflamatorios y neurodegenerativos de la enfermedad, y monitoree su respuesta al tratamiento mediante el análisis continuado de biomarcadores, es necesario para optimizar el tratamiento de la EM.
Conflicto de intereses: Los autores declaran no tener conflicto de intereses
1. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nature Reviews Neurol 2012; 8: 647-56. [ Links ]
2. Popescu BFG, Lucchinetti CF. Pathology of demyelinating diseases. Ann Rev Pathol Mechanisms of Disease 2012; 7: 185-217. [ Links ]
3. Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurology 2009; 8: 280-91. [ Links ]
4. Frohman EM, Racke MK, Raine CS. Multiple sclerosis -the plaque and its pathogenesis. N Engl J Med 2006; 354: 942-55. [ Links ]
5. Havrdova E, Galetta S, Hutchinson M, et al. Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the Natalizumab Safety and Efficacy in Relapsing-Remitting Multiple Sclerosis (AFFIRM) study. Lancet Neurol 2009; 8: 254-60. [ Links ]
6. Niino M, Bodner C, Simard ML et al. Natalizumab effects on immune cell responses in multiple sclerosis. Ann Neurol 2006; 59: 748-54. [ Links ]
7. Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006; 354: 899-910. [ Links ]
8. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010; 362: 402-15. [ Links ]
9. Kappos L, Radue EW, O'Connor P, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. New Engl J Med 2006; 355: 1124-40. [ Links ]
10. Kappos L, Radue EW, O'Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010; 362: 387-401. [ Links ]
11. Mehling M, Hilbert P, Fritz S, et al. Antigen-specific adaptive immune responses in fingolimod-treated multiple sclerosis patients. Ann Neurol 2011;69: 408-13. [ Links ]
12. O'Garra A, Murphy KM. From IL-10 to IL-12: how pathogens and their products stimulate APCs to induce T(H)1 development. Nature Immunol 2009; 10: 929-32. [ Links ]
13. Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol 2003; 21: 713-58. [ Links ]
14. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol 2009; 27: 485-517. [ Links ]
15. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med 2009; 361: 888-98. [ Links ]
16. Sie C, Korn T, Mitsdoerffer M. Th17 cells in central nervous system autoimmunity. Exp Neurol, doi:10.1016/j.expneurol.2014.03.009 (2014) (in the press). [ Links ]
17. Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1987; 1: 893-5. [ Links ]
18. Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology 1987; 37: 1097-1102 . [ Links ]
19. Deiss A, Brecht I, Haarmann A, Buttmann M. Treating multiple sclerosis with monoclonal antibodies: a 2013 update. Expert Rev Neurotherapeutics 2013; 13: 313-35. [ Links ]
20. Fernández Ó, Arnal-García C, Arroyo-González R, et al. Review of the novelties presented at the 28th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) (III). Revista de Neurología 2013; 57: 317-29. [ Links ]
21. Codarri L, Gyülvészi G, Tosevski V, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nature Immunol 2011; 12: 560-7. [ Links ]
22. El-Behi M, Ciric B, Dai H, Yan Y, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nature Immunol 2011; 12: 568-75. [ Links ]
23. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol 2007; 178: 39-48. [ Links ]
24. Peters A, Pitcher LA, Sullivan JM, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity 2011; 35: 986-96. [ Links ]
25. Quintana FJ, Farez MF, Izquierdo G, Lucas M, Cohen IR, Weiner HL. Antigen microarrays identify CNS-produced autoantibodies in RRMS. Neurology 2012; 78: 532-9. [ Links ]
26. Babbe H, Roers A, Waisman A, Lassmann H, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 2000; 192: 393-404. [ Links ]
27. Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sciences 1983; 62: 219-32. [ Links ]
28. Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol 1986; 19: 578-87. [ Links ]
29. Melzer N, Meuth SG, Wiendl H. CD8+ T cells and neuronal damage: direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. FASEB J 2009; 23: 3659-73. [ Links ]
30. Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain 2000; 123: 1174-83. [ Links ]
31. Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain 2002; 125: 2202-12. [ Links ]
32. Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science 1995; 269: 549-52. [ Links ]
33. Luessi F. Siffrin V, Zipp F. Neurodegeneration in multiple sclerosis: novel treatment strategies. Exp Rev Neurotherapeutics 2012; 12: 1061-77. [ Links ]
34. Medana IM, Gallimore A, Oxenius A, Martinic MM, Wekerle H, Neumann H. MHC class I-restricted killing of neurons by virus-specific CD8+ T lymphocytes is effected through the Fas/FasL, but not the perforin pathway. Eur J Immunol 2000; 30: 3623-33. [ Links ]
35. Giuliani F, Goodyer CG, Antel JP, Yong VW. Vulnerability of human neurons to T cell-mediated cytotoxicity. J Immunol 2003; 171: 368-79. [ Links ]
36. Baldwin RL, Stolowit, ML, Hood L, Wisnieski BJ. Structural changes of tumor necrosis factor alpha associated with membrane insertion and channel formation. Proc Natl Acad Sciences USA 1996; 93: 1021-6. [ Links ]
37. Kagan BL, Baldwin RL, Munoz D, Wisnieski BJ. Formation of ion-permeable channels by tumor necrosis factor-alpha. Science 1992; 255: 1427-30. [ Links ]
38. Venters HD, Dantzer, Kelley KW. Tumor necrosis factor-alpha induces neuronal death by silencing survival signals generated by the type I insulin-like growth factor receptor. Ann New York Acad Sciences 2000; 917: 210-20. [ Links ]
39. Venters HD, Dantzer R, Kelley KW. A new concept in neurodegeneration: TNFalpha is a silencer of survival signals. Trends Neurosciences 2000; 23: 175-80. [ Links ]
40. Mizuno T, Zhang G, Takeuchi H, et al. Interferon-gamma directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-gamma receptor and AMPA GluR1 receptor. FASEB J 2008; 22: 1797-1806. [ Links ]
41. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008; 358: 676-88. [ Links ]
42. Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol 2010; 67, 452-61. [ Links ]
43. Piccio L, Naismith RT, Trinkaus K et al. Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch Neurol 2010; 67: 707-14. [ Links ]
44. Yeste A, Quintana FJ. Antigen microarrays for the study of autoimmune diseases. Clin Chem 2013; 59: 1036-44. [ Links ]
45. Quintana FJ, Yeste A, Weiner HL, Covacu R. Lipids and lipid-reactive antibodies as biomarkers for multiple sclerosis. J Neuroimmunol 2012; 248: 53-7. [ Links ]
46. Quintana FJ, Patel B, Yeste A, et al. Epitope spreading as an early pathogenic event in pediatric multiple sclerosis. Neurology. In press. [ Links ]
47. Quintana FJ, Farez MF, Viglietta V, et al. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sciences USA 2008; 105: 18889-94. [ Links ]
48. Aslam M, Kalluri SR, Cepok S, et al. The antibody response to oligodendrocyte specific protein in multiple sclerosis. J Neuroimmunol 2010; 221: 81-6. [ Links ]
49. Chan A, Decard BF, Franke C, et al. Serum antibodies to conformational and linear epitopes of myelin oligodendrocyte glycoprotein are not elevated in the preclinical phase of muliple sclerosis. Multiple sclerosis 2010; 16: 1189-92. [ Links ]
50. Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Ann Review Immunol 2009; 27: 119-45. [ Links ]
51. Goldmann T, Prinz M. Role of microglia in CNS autoimmunity. Clin Dev Immunol 2013; 2013: 208093. [ Links ]
52. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathology 2013; 229: 176-85. [ Links ]
53. Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nature Immunol 2011; 12: 231-8. [ Links ]
54. Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurology 2001; 50: 389-400. [ Links ]
55. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Brück W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol 2013; 125: 595-608. [ Links ]
56. Diestel A, Aktas O, Hackel D, et al. Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: a link between demyelination and neuronal damage. J Exp Med 2003; 198: 1729-40. [ Links ]
57. Farez MF, Quintana FJ, Gandhi R, Izquierdo G, Lucas M, Weiner HL. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nature Immunol 2009; 10: 958-64. [ Links ]
58. Hanisch UK. Microglia as a source and target of cytokines. Glia 2002; 40: 140-55. [ Links ]
59. Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 2004; 173: 3916-24. [ Links ]
60. Takeuchi H, Jin S, Wang J, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 2006; 281: 21362-8. [ Links ]
61. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol 2010; 119: 7-35. [ Links ]
62. Brosnan CF, Raine CS. The astrocyte in multiple sclerosis revisited. Glia 2013; 61: 453-65. [ Links ]
63. Mayo L, Trauger SA, Blain M, et al. B4GALT6 controls astrocyte activation during CNS inflammation. Nat Med. In press. [ Links ]
64. Mascanfroni ID, Yeste A, Vieira SM, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nature Immunol 2013; 14: 1054-63. [ Links ]
65. Monif M, Burnstock G, Williams DA. Microglia: proliferation and activation driven by the P2X7 receptor. See comment in PubMed Commons below Int J Biochem Cell Biol 2010; 42: 1753-6. [ Links ]
66. Matute C, Torre I, Pérez-Cerdá F, et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neuroscience 2007; 27: 9525-33. [ Links ]
67. Basso AS, Frenkel D, Quintana FJ, et al. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Inv 2008; 118: 1532-43. [ Links ]
68. Mayo L, Quintana FJ, Weiner HL. The innate immune system in demyelinating disease. Immunol Rev 2012; 248: 170-87. [ Links ]
69. Boulanger JJ, Messier C. From precursors to myelinating oligodendrocytes: Contribution of intrinsic and extrinsic factors to white matter plasticity in the adult brain. Neuroscience 2014; 269C: 343-66. [ Links ]
70. Franklin RJ, Gallo V. The translational biology of remyelination: Past, present, and future. Glia. doi:10.1002/glia.22622 (2014).See comment in PubMed Commons below [Epub ahead of print] [ Links ].
71. Mi S, Pepinsky RB, Cadavid D. Blocking LINGO-1 as a therapy to promote CNS repair: from concept to the clinic. CNS drugs 2013; 27: 493-503. [ Links ]
72. Huang JK, Jarjour AA, Nait Oumesmar B, et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nature Neuroscience 2011; 14: 45-53. [ Links ]
73. Lundgaard I, Luzhynskaya A1, Stockley JH, et al. Neuregulin and BDNF induce a switch to NMDA receptor-dependent myelination by oligodendrocytes. PLoS biology 2013; 11, e1001743, doi:10.1371/journal.pbio.1001743. [ Links ]
74. Walczak A, Siger M, Ciach A, Szczepanik M, Selmaj K. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol 2013; 70: 1105-9. [ Links ]
75. Wraith DC. Therapeutic peptide vaccines for treatment of autoimmune diseases. Immunol Lett 2009; 122: 134-6. [ Links ]
76. Bar-Or A, Vollmer T, Antel J, et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch Neurol 2007; 64: 1407-15. [ Links ]
77. Garren H, Robinson WH, Krasulová E, et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol 2008; 63: 611-20. [ Links ]
78. Quintana FJ, Carmi P, Cohen IR. DNA vaccination with heat shock protein 60 inhibits cyclophosphamide-accelerated diabetes. J Immunol 2002; 169: 6030-5. [ Links ]
79. Quintana FJ, Carmi P, Mor F, Cohen IR. Inhibition of adjuvant arthritis by a DNA vaccine encoding human heat shock protein 60. J Immunol 2002; 169: 3422-8. [ Links ]
80. Quintana FJ, Carmi P, Mor F, Cohen IR. DNA fragments of the human 60-kDa heat shock protein (HSP60) vaccinate against adjuvant arthritis: identification of a regulatory HSP60 peptide. J Immunol 2003; 171: 3533-41. [ Links ]
81. Quintana FJ, Carmi P, Mor F, Cohen IR. Inhibition of adjuvant-induced arthritis by DNA vaccination with the 70-kd or the 90-kd human heat-shock protein: immune cross-regulation with the 60-kd heat-shock protein. Arthritis Rheum 2004; 50: 3712-20. [ Links ]
82. Quintana FJ, Cohen IR. DNA vaccines coding for heat-shock proteins (HSPs): tools for the activation of HSP-specific regulatory T cells. Expert opinion on biological therapy 2005; 5: 545-54. [ Links ]
83. Quintana FJ. Nanoparticles for the induction of antigen-specific Tregs. Immunotherapy 2013; 5: 437-40. [ Links ] [ Links ]
Recibido: 20-VIII-2014
Aceptado: 08-IX-2014

