










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680
Medicina (B. Aires) vol.75 no.2 Ciudad Autónoma de Buenos Aires Apr. 2015
ARTÍCULO ORIGINAL
Bases moleculares de alfa-talasemia en la Argentina
Karen G. Scheps1, Liliana Francipane2, Abigail Nash2, Gloria E. Cerrone1,Silvia B. Copelli1, Viviana Varela1
1Cátedra de Genética y Biología Molecular, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, INIGEM, CONICET-UBA,
2División Genética, Hospital de Clínicas José de San Martín, Universidad de Buenos Aires, Argentina
Dirección postal: Prof. Dr. Viviana Varela, Cátedra de Genética y Biología Molecular, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires,Junín 956, 1113 Buenos Aires, Argentina
Fax: (54-11) 4964-8296 e-mail: vvarela@ffyb.uba.ar
Recibido: 29-IX-2014
Aceptado: 12-I-2015
Resumen
La α-talasemia, es uno de los desórdenes hereditarios más frecuentes mundialmente. Al presente, el diagnóstico molecular es la única herramienta que permite el diagnóstico certero. El propósito de este trabajo fue caracterizar las bases moleculares de estos síndromes en nuestro medio, y establecer relaciones genotipo-fenotipo. Mediante la complementación de distintas técnicas de biología molecular e hibridación fluorescente in situ (FISH), se logró poner en evidencia la presencia de mutaciones α-talasémicas en 145 de 184 (78.8%) pacientes estudiados con perfil hematológico compatible con α-talasemia. Dentro de este grupo, las deleciones correspondieron al defecto genético más frecuente, prevaleciendo la mutación -α3.7 en genotipos heterocigotas y homocigotas. Asimismo, en pacientes con fenotipo α0 las deleciones prevalentes fueron –MED y –CAL/CAMP. Este estudio permitió también describir una deleción de la región sub-telomérica en un paciente con α-talasemia y retraso mental. En el 7.6% de los pacientes caracterizados clínicamente como posibles α-talasémicos (microcitosis con valores de Hb A2 inferiores al 3.5%), se hallaron mutaciones β-talasémicas en estado heterocigota. Se lograron establecer perfiles hematológicos asociados a los genotipos α+ y α0 para pacientes adultos y niños. Esperamos que este trabajo pueda servir como guía para reconocer posibles portadores α-talasémicos. También permite destacar el trabajo en conjunto de médicos hematólogos, el laboratorio (bioquímico y de biología molecular) y de los médicos genetistas, con el fin de proporcionar adecuado consejo genético.
Palabras clave: Alfa-talasemia; Genética; Deleción; Mutación puntual; Biología molecular.
Abstract
Molecular bases of α-thalassemia in Argentina. The α-thalassemia is one of the most common hereditary disorders worldwide. Currently, molecular diagnostics is the only available tool to achieve an accurate diagnosis. The purpose of this study was to characterize the molecular bases of these syndromes in our environment and to establish genotype-phenotype associations. Through a combination of different molecular techniques and fluorescent in situ hybridization (FISH),we were able to find α-thalassemic mutations in 145 of the 184 patients (78.8%) studied with hematological parameters compatible with α-thalassemia. Deletions of the a-globin genes resulted the major molecular cause of the disease, and the most frequent mutation was -α3.7, found in homozygous and heterozygous genotypes. In patients with α0 phenotypes, other prevalent mutations were –MED and –CAL/CAMP. The description of a sub-telomeric deletion in a patient with α-thalassemia and mental retardation was also achieved. β-thalassemic mutations in heterozygous state were found in 7.6% of the patients, who presented α-thalassemic clinical features (microcytosis and Hb A2 levels below 3.5%). Hematologic profiles for the α+ and α0 genotypes were established for adult and pediatric patients. Hopefully, this work will provide guidelines for the detection of possible α-thalassemic carriers. It also highlights the collaborative work of hematologists, the biochemical and molecular biology laboratory and genetists, in order to provide appropriate genetic counseling.
Key words: Alpha-thalassemia; Genetics; Deletion; Puntual mutation; Molecular biology.
La a-talasemia (a-tal) (OMIM 604131; HBA2: 141850, HBA1: 141800) es uno de los desórdenes genéticos monogénicos más comunes a nivel mundial. Se estima que el 5% de la población mundial es portadora de alguna mutación α-tal1. Prevalece en regiones tropicales y sub-tropicales (el sub-Sahara africano, Mediterráneo, Oriente Medio, el subcontinente indio y el sudeste de Asia2), coexistiendo en regiones donde está difundida la malaria, que ejerció una presión evolutiva positiva sobre las mutaciones α-tal. Cabe destacar que en varias zonas de China y del sudeste asiático, el 40% de la población es portadora de alelos α-tal3.
Los genes HBA2 y HBA1, que codifican las cadenas de α-globina, se encuentran en la región telomérica del brazo corto del cromosoma 16 (16p13.3); en el cluster (o familia de genes) de α-globina que incluye: el gen embrionario HBZ, el pseudogen HBZP, el gen HBM, el pseudogen HBAP1, los genes HBA2 y HBA1 y HBQ1.
La expresión tejido-específica de los genes del cluster alcanza altos niveles a través de interacciones de largo alcance entre secuencias distales regulatorias y los promotores. Los elementos distales se encuentran altamente conservados evolutivamente, razón por la cual se los conoce como MCS (Multispecies Conserved Sequences) o MRE (Major Regulatory Element) por su rol en la transcripción. A pesar de existir 4 secuencias MCS (-R1 a -R4), la única secuencia que hasta el momento demostró cumplir el verdadero rol de enhancer es MCS-R2, también conocida como HS-40, por ubicarse a 40 kb corriente arriba del cluster.
Los síndromes α-tal presentan gran heterogeneidad fenotípica, producto de la diversidad de sus bases moleculares. La causa más frecuente de las α-tal son las deleciones, con la pérdida de una (-a) o ambas (--) copias de los genes HBA de un mismo cromosoma. En función del número de genes afectados, los portadores α-tal se clasifican en: "portador silente", con 3 copias de HBA funcionantes (genotipo -α/αα) clínicamente normal y que raramente cursa con anemia, aunque exhibe ciertas alteraciones en el hemograma, el portador con "rasgo talasémico", que posee 2 copias funcionantes de genes que codifican para α-globina (genotipos -α/-α o --/α α) y dos presentaciones clínicamente relevantes: la enfermedad de hemoglobina (Hb) H, (genotipo -α/--) y el síndrome de hidropesía fetal por Hb Bart, (genotipo --/--)4. En la enfermedad de la Hb H, el exceso de cadenas β, forman homotetrámeros (β4) que, a medida que maduran los glóbulos rojos, precipitan formando cuerpos de inclusión. En estos individuos, los efectos clínicos de la anemia son exacerbados porque los tetrámeros b4 no pueden ceder oxígeno a los tejidos5. Las manifestaciones clínicas más importantes son la anemia hemolítica, que puede llegar al requerimiento de transfusiones, hepatoesplenomegalia e ictericia. También pueden presentar modificaciones óseas producto de eritropoyesis extra-medular.
El síndrome de hidropesía fetal por Hb Bart es la condición más grave y es fundamentalmente incompatible con la vida extrauterina, ya que los individuos afectados son incapaces de producir Hb F, Hb A o Hb A2; es relativamente común en el sudeste asiático y es menos frecuente en la población de origen mediterráneo.
Se describieron además, formas no clásicas de presentación de α-tal asociada a retraso mental y α-tal adquirida en síndromes mielodisplásicos. Dentro de los pacientes con retraso mental, que en general cursan con enfermedad de Hb H, existen 2 grupos de pacientes: el primero, con retraso mental relativamente leve, variedad de dismorfias y retraso en el desarrollo, es catalogado como síndrome ATR 16. A nivel molecular corresponde a un síndrome de deleción contiguo en la región telomérica del brazo corto del cromosoma 16, generalmente esporádico, que conduce a la pérdida de los genes del cluster de α-globina y genes adyacentes; hasta el momento no se pudo esclarecer cuáles están involucrados en esta condición6. El segundo grupo de pacientes presenta alteraciones clínicas graves con compromiso neurológico (relativamente uniformes: signos dismórficos característicos como hipertelorismo, puente nasal deprimido, narinas antevertidas, boca grande en carpa, anomalías genitales, hipotonía y retraso mental grave, debido a mutaciones a nivel germinal en el gen ATRX. La α-tal asociada a los síndromes mielodisplásicos también se atribuye a mutaciones adquiridas en el gen ATRX7.
Las deleciones que afectan el cluster α presentan un patrón de distribución geográfica definido. Prevalecen las que provocan la pérdida de una sola copia de gen α (del α+) y son producto de crossing-over desigual entre regiones de alta homología en el cluster de α-globina. La de mayor frecuencia a nivel mundial es la deleción -α3.7o rightward deletion (deleción de 3.7 kb) que predomina en el área del Mediterráneo, África y Asia. La deleción -a4.2 o leftward deletion (deleción de 4.2 kb) es menos frecuente y prevalece en el sudeste asiático8.
Hay descriptas más de 40 deleciones α04, con la pérdida de ambas copias de genes HBA yque pueden incluir otros genes del cluster. Las más frecuentes son --MEDI y -(α)20.5 en población del Mediterráneo, --SEA en el sudeste de Asia, y --FIL en población filipina.
Con menor frecuencia, las bases moleculares de las α-tal están asociadas a mutaciones puntuales o no delecionales. Prevalecen las alteraciones en el gen HBA2 (αMα) sobre el gen HBA1 (αα M)8, aunque es posible que exista un sub-diagnóstico de alteraciones en el último gen. Las más frecuentes en el área del Mediterráneo son αNcoIα (HBA2:c.2T>C: cambio ATG > ACG en el codón de iniciación) y αIVSI(-5nt)α (HBA2:c.95+2_95+6delTGAGG: eliminación de 5 nucleótidos en la secuencia dadora de splicing del intrón 1 de HBA2). Se han informado otras mutaciones: alteraciones en la señal de poliadenilación, descriptas en el Mediterráneo y Oriente Medio, y en el codón de terminación que conducen a variantes elongadas de Hb, como Hbs Constant Spring, Icaria, Koya Dora, y Seal Rock Paksé, descriptas en el área del Mediterráneo y en Asia9.
Los desórdenes hereditarios que afectan la síntesis normal de Hb son comunes en Argentina, siendo la β-tal la alteración más frecuente y mejor caracterizada10-12. No obstante, los patrones migratorios están cambiando dramáticamente la distribución geográfica mundial de α-tal, al punto de incluirse en el panel de screening neonatal obligatorio en el estado de California y contemplarse su inclusión a nivel nacional en EE.UU.13.
Al presente, el diagnóstico molecular es la única herramienta que permite el diagnóstico certero y la diferenciación de anemias causadas por otras etiologías. No existen marcadores clínicos o hematológicos confirmatorios de α-tal y la información reportada sobre las bases moleculares de estos síndromes en nuestro medio es escasa y parcial14, 15.
El propósito de este trabajo fue caracterizar las alteraciones genéticas en el cluster de α-globina asociadas a fenotipos compatibles con α-tal y establecer la relación entre el genotipo y el fenotipo hematológico.
Materiales y métodos
Se derivaron para análisis molecular, a nuestro laboratorio, 184 pacientes con fenotipo hematológico compatible con a-tal. Previo a los estudios, los pacientes firmaron un consentimiento informado que contempla la Declaración de Helsinki de la Asociación Médica Mundial. Se purificó el ADN genómico de leucocitos de sangre periférica por el método de bromuro de cetiltrimetilamonio (CTAB)16. La estrategia de estudio molecular comprendió, en principio, la detección e identificación de las deleciones -α3.7, -α4.2,--MEDI, -(α)20.5, --SEA y --FIL por PCR GAP17. Se diseñaron primers para la detección de la deleción --SPAN por PCR GAP y para la búsqueda de otras deleciones se utilizó la técnica de Multiplex ligation-dependent-probe-amplification (MLPA) mediante el kit "SALSA MLPA probemix P140-B4 HBA", MRC-Holland.
En los pacientes en que no se hallaron deleciones, se estudió la presencia de mutaciones puntuales, en HBA2 y HBA1. Se amplificaron diferencialmente los genes HBA2 y HBA1 con primers específicos18. Las mutaciones αNcoIα y αIVSI(-5 nt)α se estudiaron por PCR y posterior digestión enzimática. Otras variaciones de secuencia se identificaron por secuenciación automática en un ABI PRISM™ 3130XL (Applied Biosystems, Seoul, Korea). Cuando se encontraron mutaciones puntuales no descriptas previamente, se caracterizaron por clonado con el sistema pGEM ®-T Easy Vector System (Promega, Madison, WI, USA), transformación de bacterias Escherichia coli DH5α y secuenciación de los clones obtenidos. En un grupo de pacientes se estudió el gen HBB por PCR y secuenciación con los primers HBB1-F and HBB2-R10 y los primers L3 5´- GGGTACAGTTTAGAATGGG-3´ y HBB4-R: 5'-TAGCTGTTTGCAGCCTCACCTT-3' diseñados en nuestro laboratorio (PrimerQuest ®, IDT, Coralville, USA). Los análisis bioinformáticos de las secuencias se realizaron con los programas Finch-TV Version 1.4.0 (Geospiza Inc., USA), ChromasPro 1.32 (Technelysium, PtyLtd, Australia) y BLAST19.
La información sobre las mutaciones estudiadas se obtuvo a partir de la base de datos "A Database of Human Hemoglobin Variants and Thalassemias" (http://globin.cse.psu.edu/globin/hbvar/menu.html), y del GenBank se tomó la secuencia normal de referencia de los genes (HBA: NC_000016.10 y HBB:NC_000011.10 ).
Las diferencias entre los valores hematimétricos de los distintos grupos se analizaron mediante One-way ANOVA y el test post-hoc de comparaciones múltiples de Bonferroni realizados con GraphPadPrism version 5.03 para Windows, GraphPad Software (San Diego California USA, www.graphpad.com).
Resultados
En 145 pacientes se hallaron mutaciones α-tal, responsables del fenotipo clínico-hematológico. En la Tabla 1 se enumeran las distintas mutaciones halladas y su distribución. La deleción -α3.7 es la que presentó mayor frecuencia siendo la principal responsable, tanto del fenotipo "portador silente" (genotipo -α3.7/αα), como del "rasgo talasémico" (genotipo -α3.7/-α3.7).
Tabla 1. Distribución de las mutaciones α-tal halladas en los pacientes estudiados
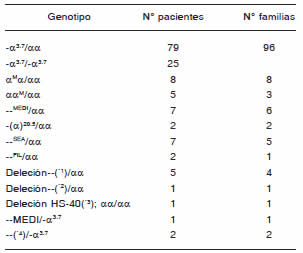
Las deleciones indicadas como (*) fueron caracterizadas por MLPA: (*1): --CALo--CAMPANIA; (*2):deleción entre 10.2 y 14.2 kb; (*3):deleción entre 0.1 y 89.6 kb;(*4): deleción mayor a 127.5 kb.
Se encontraron distintas mutaciones α0 en estado heterocigota: la deleción --MEDI resultó ser la más frecuente en pacientes con ascendencia mediterránea, y la deleción --SEA en los de origen asiático. La mutación --MEDI se halló también asociada a la deleción -α3.7 en un paciente con enfermedad de Hb H.
En 13 pacientes, con fenotipo compatible con α0-tal, en que no se pudieron caracterizar las mutaciones por las estrategias antes mencionadas (11 con fenotipo de "rasgo talasémico" y 2 con Hb H, positivos para la deleción -α3.7), se realizó el análisis por MLPA (amplificación múltiple de sondas dependientes de ligación). Por medio de esta técnica se pudo confirmar una alteración en el cluster de α-globina como el defecto responsable del fenotipo en 9 pacientes. En 5 de ellos (4 familias) se determinó la presencia de una deleción que comprende entre 28.9 y 36.5 kilo-pares de bases (kb), que sería compatible con las deleciones --CAL (cuyos límites fueron actualizados recientemente20) o --CAMPANIA 21, muy similares en tamaño y ambas caracterizadas en población del Mediterráneo. En un paciente se identificó una deleción cuyos bordes no coincidirían con otras deleciones previamente descriptas, con un tamaño entre 10.2 y 14.2 kb, que involucra la pérdida de ambos genes HBA. En otro se observó la deleción de la región regulatoria HS-40 de entre 0.1 y 89.6 kb en uno de sus alelos, mientras que sus 4 copias de genes HBA permanecieron intactas. En los 2 pacientes con Hb H en los que únicamente se había identificado la deleción -α3.7, el análisis por MLPA reveló la presencia de grandes deleciones (mayores a 127.5 kb) que abarcaron todas las sondas incluidas en el kit. Uno de estos pacientes presentó, además de la α-tal, retraso mental y anomalías en el desarrollo. Los restantes 4 pacientes, con fenotipo compatible con α0-tal, no presentaron deleciones ni inserciones por MLPA. En la Fig. 1 se esquematizan los resultados obtenidos por MLPA.
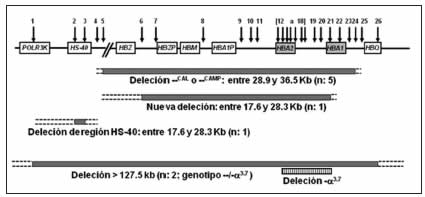
Fig. 1. Esquema de las deleciones caracterizadas por Multiplex ligation-dependent-probe-amplification (MLPA)
Las flechas indican la posición de las 26 sondas incluidas en el kit de MLPA. Las diferentes deleciones observadas en pacientes se indican como barras grises; las líneas punteadas marcan las regiones de los posibles puntos de corte de la deleción. En los 2 pacientes con Hb H (genotipo --/-α3.7), se indican las 2 deleciones: la deleción caracterizada por MLPA que se extendió a lo largo de las 26 sondas (barras grises) y la deleción -α3.7 (barras verticales). n: número de pacientes en los que se encontraron las mutaciones
En los pacientes en que no se hallaron deleciones, se estudió la presencia de mutaciones puntuales en HBA2 y HBA1. En 8 familias se hallaron mutaciones en estado heterocigota en el gen HBA2: 5 presentaron la mutación αIVSI(-5 nt)α, 2 la αNcoIα y, en uno, una variante de secuencia en la región 3' UTR, HBA2:c.*107. Estos hallazgos son compatibles con el perfil étnico de nuestra población, de ascendencia predominante del área del Mediterráneo. En 3 familias, se hallaron mutaciones en el gen HBA1 previamente no descriptas: en una paciente adulta se determinó una sustitución en la secuencia aceptora de splicing, HBA1:c.301-2A>T, que se detectó en cis con el cambio que da lugar a la Hb Riccarton22. Por otra parte, se encontraron dos deleciones noveles que generan corrimientos del marco de lectura: HBA1:c.187delG (p.W62fsX66), en una mujer con anemia drepanocítica y su hijo portador de Hb S, en asociación con la variante patchwork α21223 y la mutación HBA1:c.237de lC (p.Asn78Lysfs*6) en un padre y su hijo, de la última familia.
En la población estudiada no se encontraron las mutaciones -α4.2, --SPAN, ni otras mutaciones no delecionales.
A partir de los resultados obtenidos, se correlacionaron las mutaciones halladas con los parámetros hematológicos. En la Tabla 2 se presentan los resultados hallados para el recuento de glóbulos rojos (Rto.GR), volumen corpuscular medio (VCM), hemoglobina corpuscular media (HCM) y Hb A2. Los pacientes se agruparon de acuerdo a su genotipo, sexo y rango etario, con el fin de evaluar si existían diferencias significativas en los valores entre estos grupos. Tanto para el VCM como el HCM, en el genotipo -α/αα no existen diferencias significativas entre hombres y mujeres adultos, pero sí entre pacientes adultos y niños, mientras que en los genotipos -α/-α y --/αα no se observaron diferencias significativas entre los distintos grupos. Para el recuento de glóbulos rojos no se observaron diferencias significativas entre los grupos para el genotipo -α/αα, pero en el caso de los pacientes con deleción de 2 copias de genes HBA, los hombres presentaron valores significativamente más elevados que las mujeres y que los niños. El análisis de los valores de Hb A2 arrojó resultados similares a los observados para el VCM y el HCM, con la salvedad de que los valores remitidos fueron obtenidos por distintos laboratorios, por lo que la diferencia significativa entre adultos y niños con el grupo -α/αα podría ser atribuida a esta causa.
Tabla 2. Relación entre los genotipos α-tal y los parámetros hematimétricos

VCM: volumen corpuscular medio; HCM: hemoglobina corpuscular media. Se indican media ± desvío estándar para cada parámetro.
Al comparar el VCM y HCM entre los distintos genotipos (en este caso se incluyeron las mutaciones puntuales, sin discriminar entre el gen afectado y sin ajuste por sexo y edad), se hallaron diferencias significativas en los valores presentados por los distintos genotipos (-α/αα, portadores de mutaciones puntuales y de doble deleción).
Los valores de VCM arrojaron diferencias significativas para los distintos genotipos (-α/αα, portadores de mutaciones puntuales o de deleción de dos copias de genes HBA). No se registraron diferencias significativas, únicamente entre los niños con deleción de una copia de gen HBA y aquellos que presentaron mutaciones puntuales.
En el análisis de la HCM, los resultados fueron bastante similares a los del VCM, excepto que tampoco en adultos se verificaron diferencias significativas en los valores de los portadores del genotipo -α/αα y de mutaciones puntuales. Las tendencias en los valores analizados se representan en la Fig. 2.
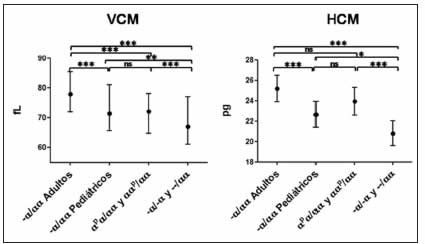
Fig 2. Comparación de VCM y HCM entre los distintos grupos
Valores de VCM y HCM están indicados como media ± DS. Análisis estadístico comparativo de VCM y HCM de los distintos grupos por ANOVA de una vía y test post-hoc de Bonferroni (GraphPadPrism versión 5.03). Valores de significancia: ***p = 0.001; **p = 0.01; *p = 0.05; ns: no significativo.
En un grupo de 14 pacientes en los que no se encontraron mutaciones con Hb A2 ≤ a 3.5% (rango de 2.4% a 3.4%) y VCM < 75 fl, se secuenció el gen HBB y se identificaron mutaciones b-tal en estado heterocigota, siendo IVS 1-6 (T > C) la responsable del cuadro clínico-hematológico en la mitad de los casos (el resto de las mutaciones fueron Codón 39 (C > T), IVS 1-110 (G > A) y (-87) (C > G). Solo un paciente presentó niveles de Hb F mayores al 2.0%, que podrían justificar una menor producción de Hb A2.
En 25 pacientes (13.6% de los casos), 11 con fenotipo compatible con α+-tal y 14 con α0-tal, hasta el momento no se halló el defecto primario y continúan en estudio.
Discusión
Los resultados obtenidos muestran que la deleción -α3.7 es la más frecuente en nuestro medio, al igual que lo descripto en la mayoría de las poblaciones analizadas. Esto implica que ante la sospecha de α-tal, ésta debe ser la primera mutación a identificar. En pacientes con "rasgo talasémico", negativo para la deleción -α3.7, es importante conocer su ascendencia debido a que las mutaciones --MEDI y -(α)20.5, solo fueron halladas en individuos con ascendencia mediterránea, y las deleciones --SEA y --FIL, en individuos de origen asiático.
A partir del estudio por MLPA se observó en 5 pacientes (4 familias genéticamente no relacionadas), de ascendencia mediterránea, el mismo patrón de deleción (compatible con --CAL o --CAMPANIA) lo que convertiría a esta alteración en la tercera mutación más frecuente en nuestra población, luego de las deleciones -α3.7 y --MEDI. Actualmente estamos trabajando para establecer los límites de esta deleción, y diseñar un ensayo directo para su identificación.
La deleción de la región regulatoria MCS-R2 (HS-40) tiene la misma repercusión en el fenotipo hematológico que la pérdida de 2 copias de HBA, resaltando la importancia de la presencia de esta región para la expresión en niveles apropiados de los genes del cluster α.
En las familias tipificadas, las mutaciones puntuales representaron el 8.5% del defecto genético responsable; el hecho de haber descripto 3 mutaciones noveles en HBA1 destaca la importancia de incluir la secuenciación de los genes HBA en el estudio de pacientes con fenotipos α-tal en los que se descartaron las deleciones y mutaciones puntuales más frecuentes.
De las formas clínicamente relevantes de α-tal, solo ingresaron para su estudio 3 pacientes con Hb H y, hasta el momento, no registramos hidropesía fetal por Hb Bart en la población estudiada. Este panorama podría cambiar debido a un incremento en la inmigración asiática de las últimas décadas, que presenta mayor prevalencia de rasgo α-tal y bases genéticas diferentes. En los 3 pacientes con Hb H, ninguno de ascendencia oriental, se determinó que la base molecular correspondía a la deleción de 3 copias de HBA: en un caso el genotipo fue -α3.7/--MEDI y en los otros dos, -α3.7/deleción ≥ 127.5 kb (deleción caracterizada por MLPA). El hallazgo del defecto molecular en las Hb H es de suma importancia, ya que en los pacientes que presentan una mutación puntual, el cuadro suele ser más grave24.
En el paciente con Hb H y retraso mental asociado, se realizó un estudio de cariotipo, en el que no se visualizaron alteraciones, y un estudio de FISH, en el que pudo ponerse en evidencia la deleción subtelomérica. La presencia de esta deleción del cluster de α-globina y genes adyacentes podría estar asociada o contribuir al fenotipo observado. Este caso demuestra la importancia de la búsqueda de deleciones crípticas en pacientes con retraso mental, y reitera la necesidad del estudio molecular en pacientes en que el retraso se presente con anemia microcítica e hipocrómica con niveles de hierro normales.
Es interesante destacar el porcentaje de pacientes que presentaron mutaciones b-tal y fueron derivados con diagnóstico presuntivo de α-tal. El hecho de que en el 50% de los casos la mutación responsable fue IVS 1-6 (mutación tipo b+ leve) indicaría que el déficit de cadenas de β-globina, producto de esta alteración, no siempre sería lo suficientemente importante como para inducir aumentos de Hb A2 mayores a 3.5%. Otra observación destacable es que ninguna de las muestras en las que se hallaron mutaciones α-tal presentó niveles de Hb A2 mayores al 3.0%.
Las mutaciones α-tal presentan perfiles hematológicos definidos: el recuento de glóbulos rojos es mayor o igual a 4.5 106/mm3, el VCM suele ser menor a 80 ƒ l y la HCM es menor a 27 pg. Asimismo, se distinguen perfiles distintos para las mutaciones α+ y α0, que pueden orientar al diagnóstico; estos hallazgos demuestran que la clave para la detección y caracterización exitosa de las hemoglobinopatías, en particular las talasemias, es la minuciosa recolección inicial de los datos clínico-hematológicos de los pacientes. Finalmente, los resultados obtenidos destacan la importancia del diagnóstico molecular como herramienta fundamental para establecer un diagnóstico definitivo de estos cuadros, y el correcto asesoramiento genético-familiar.
Agradecimientos: Este trabajo fue parcialmente financiado con el subsidio 20020120200092BA otorgado por la Universidad de Buenos Aires (Programación Científica 2013-2015). Agradecemos a los pacientes y los médicos hematólogos que depositaron su confianza en nuestro trabajo.
Conflicto de intereses: Ninguno para declarar
1. Weatherall DJ. Hemoglobinopathies worldwide: present and future. Curr Mol Med 2008; 8: 592-9. [ Links ]
2. Higgs DR, Weatherall DJ. The alpha thalassaemias. Cell Mol Life Sci 2009; 66: 1154-62. [ Links ]
3. Vichinsky E. Advances in the treatment of alpha-thalassemia. Blood Rev 2012; 26 Suppl 1: S31-4. [ Links ]
4. Galanello R, Cao A. Gene test review. Alpha-thalassemia. Genet Med 2011; 13: 83-8. [ Links ]
5. Olivieri NF, Weatherall DJ. Thalassemias. In: RJ Arceci, IM Hann, OP Smith (eds) Pediatric Hematology. 3rd ed. Oxford, UK: Blackwell Publishing Ltd. 2006, p 281-301.
6. Higgs DR. The molecular basis of α-thalassemia. Cold Spring Harb Perspect Med 2013; 3:a011718.doi: 10.1101/cshperspect.a011718.
7. Galanello R, Cao A. Gene test review.Alpha-thalassemia. Genet Med 2011; 13: 83-8. [ Links ]
8. Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis 2010; 28: 5-13. [ Links ]
9. Higgs DR. The molecular basis of alpha-thalassemia. In: Steinberg MH, Forget BG, Higgs DR, Watherall DJ (eds); Disorders of hemoglobin: genetics, pathophysiology and clinical management. 2nd ed. Cambridge, UK: Cambridge University Press. 2009, p 241-65.
10. Rossetti LC, Targovnik HM, Varela V. The molecular basis of ß-thalassemia in Argentina. Influence of the immigration pattern from the Mediterranean Basin, Haematologica 2004; 89: 746-7. [ Links ]
11. Rossetti LC, Scheps KG, Binaghi A, Abreu MS, Mansilla MT, Varela V. Diagnóstico molecular de mutaciones beta talasémicas, genotipos complejos. Hematología Argentina 2010; 14: 41-7. [ Links ]
12. Torres FA, Bonduel M, Sciuccati G, et al. B Talasemia mayor en Argentina. Medicina (B Aires) 2002; 62: 124-34. [ Links ]
13. Michlitsch J, Azimi M, Hoppe C, et al. Newborn screening for hemoglobinopathies in California. Pediatr Blood Cancer 2009; 52: 486-90. [ Links ]
14. Noguera NI, Bragós IM, Milani AC. Prevalence of -alpha 3.7-thalassemia in Argentina. Hemoglobin 2002; 26: 103-6. [ Links ]
15. Bragós IM, Noguera NI, Raviola MP, Milani AC. Triplication (/alphaalphaalpha anti3.7) or deletion (-alpha3.7/) association in Argentinian beta-thalassemic carriers. Ann Hematol 2003; 82: 696-8. [ Links ]
16. Murray MG, Thompson WF. Rapid isolation of high molecular-weight plant DNA. Nucleic Acids Res 1980; 8: 4321-5. [ Links ]
17. Tan AS, Quah TC, Low PS, Chong SS. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood 2001; 98: 250-1. [ Links ]
18. Jorge SB, Melo MB, Costa FF, Sonati MF. Screening for mutations in human alpha-globin genes by nonradioactive single-strand conformation polymorphism. Braz J Med Biol Res 2003; 36: 1471-4. [ Links ]
19. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215: 403-10. [ Links ]
20. Blattner A, Brunner-Agten S, Ludin K, et al. Detection of germline rearrangements in patients with α- and β-thalassemia using high resolution array CGH. Blood Cells Mol Dis 2013; 51: 39-47.
21. Sessa R, Puzone S, Ammirabile M, et al. Identification and molecular characterization of the --CAMPANIA deletion, a novel α0-thalassemic defect, in two unrelated Italian families. Am J Haem 2010; 85: 143-4.
22. Scheps KG, Binaghi A, Varela V. Identification of a new HBA1 gene mutation (HBA1:c.301-2A>T) in cis with HbRiccarton (HBA1:c.154G>A) [α51(CE9)Gly→Ser]. Hemoglobin 2012; 36: 504-7.
23. Scheps KG, De Paula SM, Bitsman AR, et al. Coinheritance of a novel mutation on the HBA1 gene: c.187delG(p.W62fsX66) [codon 62 (-G) (α1)] with the α212 patchwork allele and Hb S[β6(A3)Glu→Val, GAG>GTG; HBB: c.20A>T]. Hemoglobin 2013; 37: 492-500.
24. Vichinsky EP. Clinical manifestations of α-thalassemia. Cold Spring Harb Perspect Med 2013; 3: a011742.

