










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680
Medicina (B. Aires) vol.75 no.4 Ciudad Autónoma de Buenos Aires ago. 2015
ARTÍCULO ESPECIAL
Hemofilia adquirida (inhibidor adquirido del factor VIII)
José M. Ceresetto1, Cristina Duboscq1, Carlos Fondevila2, Miguel Tezanos Pinto3
1Servicio de Hematología y Hemoterapia, Hospital Británico de Buenos Aires,
2Servicio de Hematología, Sanatorio Bazterrica,
3Instituto de Investigaciones Hematológicas Mariano R. Castex, Academia Nacional de Medicina, Buenos Aires, Argentina
Dirección postal: Dr. José M Ceresetto, Servicio de Hematología y Hemoterapia, Hospital Británico de Buenos Aires, Perdriel 74, 1280 Buenos Aires, Argentina
e-mail: jceresetto@intramed.net
Recibido: 30-IV-2015
Aceptado: 17-VII-2015
Resumen
La hemofilia adquirida es una enfermedad de muy poco frecuente presentación. El paciente habitualmente consulta con equimosis y hematomas extensos en la piel y tejido celular subcutáneo, anemia y en algunas oportunidades un sangrado grave, que si no se controla puede ser fatal hasta en el 20% de los casos. Se produce por un autoanticuerpo dirigido contra el factor VIII de la coagulación y suele ocurrir en pacientes añosos sin historia de sangrados, pero también puede presentarse asociado a neoplasias, enfermedades autoinmunes, medicamentos y en mujeres jóvenes asociado al embarazo. Tiene un perfil de laboratorio característico con un tiempo de tromboplastina parcial activada (aPTT) prolongado, que no corrige con plasma normal, y niveles de factor VIII disminuidos. El tratamiento recomendado es muy específico, ya que para controlar el sangrado se utilizan agentes de puenteo (productos que sortean el efecto del inhibidor), factor VII recombinante activado o concentrado de complejo de protrombina activada, y medicación inmunosupresora para erradicar el autoanticuerpo.
Palabras clave: Hemofilia adquirida; Inhibidor del factor VIII; Sangrado.
Abstract
Acquired haemophilia (acquired factor VIII inhibitor)
Acquired haemophilia is a rare disorder. The clinical picture ranges from mild ecchymosis and anaemia to life threatening bleeding in up to 20% of patients. The disease is produced by an antibody against Factor VIII and it usually occurs in the elderly, with no previous history of a bleeding disorder. It can be associated to an underlying condition such as cancer, autoimmune disorders, drugs or pregnancy. It has a typical laboratory pattern with isolated prolonged activated partial thromboplastin time (aPTT) that fails to correct upon mixing tests with normal plasma and low levels of factor VIII. Treatment recommendations are based on controlling the acute bleeding episodes with either bypassing agent, recombinant activated factor VII or activated prothrombin complex concentrate, and eradication of the antibody with immunosuppressive therapy.
Key words: Acquired hemophilia; Factor VIII inhibitor; Bleeding.
La “hemofilia adquirida” (HA) o inhibidor adquirido contra el factor VIII (FVIII) es una enfermedad autoinmune infrecuente que se presenta como un sangrado súbito y grave en pacientes sin historia de coagulopatía previa. Se trata de una enfermedad hemorragípara producida por un autoanticuerpo específico contra el Factor VIII. Este anticuerpo tiene un comportamiento crónico por su persistencia en el tiempo y un alto riesgo de recaídas1, 2. Su incidencia es de 1.5 casos cada millón de habitantes por año, pero entre los 65 y 85 años llega a ser de 9 casos por millón de habitantes y en los mayores de 85 años, 15 por millón3. Sin bien tiene mayor prevalencia en pacientes añosos y de sexo masculino, también puede presentarse en mujeres jóvenes durante el embarazo y post parto, o con enfermedades autoinmunes. De hecho, como la población anciana es la que más ha crecido en los últimos años, es de esperarse un aumento en la prevalencia de esta enfermedad en un futuro cercano4.
Tiene una alta mortalidad asociada al sangrado y a la toxicidad del tratamiento. La tasa de mortalidad por sangrado se encuentra entre 20 y 30%, llegando a ser ésta mayor a 40% si el paciente no recibe ningún tratamiento5. Un meta análisis reciente sitúa la mortalidad por sangrado entre 10 y 20%, aun en los países desarrollados7. El registro EACH2 (European Acquired Hemophilia Registry) tuvo como objetivo obtener, en forma prospectiva, datos sobre las características demográficas, diagnóstico, patologías subyacentes, presentación clínica, tratamiento y evolución de los pacientes con HA. Entre las conclusiones, merece mencionarse que la mortalidad asociada a sangrado en este registro Europeo fue de apenas 4.5%. Sin embargo, no ha sido posible corroborar estos datos en otros registros. Y, por otro lado, otros estudios publicados señalan que la mortalidad global en esta enfermedad estaría alrededor de 33% al año del diagnóstico8.
El objetivo de este trabajo es hacer una actualización sobre hemofilia adquirida teniendo en mente al médico general, internista o de cuidados críticos, quienes habitualmente serán los primeros en asistir al paciente. Otro objetivo es brindar al especialista en hematología una actualización que permita seleccionar el tratamiento más adecuado para esta rara y potencialmente fatal enfermedad.
Cuadro clínico
Al momento del diagnóstico, más del 90% de los episodios de sangrado son graves y requieren internación. Por esto, la hemofilia adquirida debe contar con un reconocimiento temprano y un diagnóstico rápido para permitir el tratamiento de la hemorragia, evitando procedimientos invasivos potencialmente peligrosos mientras se busca erradicar al inhibidor3, 5. Es muy importante realizar la consulta con un hematólogo con experiencia en inhibidores y contar con un laboratorio especializado en hemostasia. Un diagnóstico tardío puede aumentar la mortalidad de esta afección9.
El sangrado no se asemeja al de la hemofilia congénita, más bien parece el típico del paciente con un defecto en la hemostasia primaria3. Suele ser extenso y afectar a piel, mucosas y tejido celular subcutáneo en forma de equimosis o hematomas muy característicos que, cuando ocurren en los miembros inferiores o superiores, pueden producir un síndrome compartimental por isquemia secundaria a compresión de tejidos6. También se puede presentar como hematomas musculares espontáneos o ante traumas mínimos, o como una hemorragia digestiva, pulmonar, sangrado post parto o del sistema nervioso central (SNC). En general se acompaña de anemia (hemoglobina < 8 g/dl o caída > 2 g/dl que requiere transfusiones). A diferencia de la hemofilia congénita, habitualmente no presenta hemartrosis. Dado que el sangrado en algunas oportunidades es interno, se deberá monitorear con imágenes seriadas en caso de colecciones situadas en sitios no visibles11.
El diagnóstico diferencial incluye aquellos cuadros que se presentan con extenso sangrado de piel y partes blandas, como por ejemplo: coagulación intravascular diseminada crónica para-neoplásica, hiperfibrinolisis, púrpura vasculítica o amiloide, así como el sangrado asociado al tratamiento con fármacos anticoagulantes.
Estas son imágenes típicas en pacientes con hemofilia adquirida con grandes hematomas y sangrados importantes, que pueden asociarse a isquemia secundaria a compresión de tejidos y anemia (Fig. 1).

Fig. 1. Hematoma en flanco y dorso en paciente con hemofilia adquirida.
La HA suele asociarse a otras enfermedades en la mitad de los casos (15% enfermedades autoinmunes, como lupus eritematoso sistémico y artritis reumatoidea; 15% cáncer, siendo los más frecuentes de próstata y pulmón, además de los síndromes linfoproliferativos; 10% embarazo y postparto; 3-5% fármacos). Sin embargo, en el otro 50% no tiene ninguna enfermedad subyacente y se lo considera como un fenómeno autoinmune primario4-10.
Cuando el inhibidor se asocia al embarazo, generalmente se presenta entre 2 y 5 meses post parto y rara vez da síntomas durante el embarazo. Habitualmente ocurre en primíparas y no reaparece en los embarazos subsiguientes. Suele tener un curso benigno, con un bajo título de inhibidor y el anticuerpo desaparece espontáneamente en las dos terceras partes de los casos12. Al tratarse de un anticuerpo IgG, que podría eventualmente pasar al feto por vía transplacentaria y provocar sangrado, debemos supervisar muy cuidadosamente a la mujer embarazada con inhibidor, aunque las hemorragias fetales son excepcionales.
Por tratarse la HA de una enfermedad infrecuente, la experiencia y reporte de casos y los estudios prospectivos son escasos. Si combinamos todos los registros internacionales publicados, apenas se han evaluado en forma sistemática hasta la fecha menos de 1200 pacientes con esta enfermedad (Tabla 1). En la Argentina el mayor registro de HA publicado es retrospectivo e incluyó 27 pacientes entre 1991 y 2013, con un promedio de edad de 59 años, 18 (66%) eran mujeres y en 15% se asoció a embarazo. Solo un paciente falleció por sangrado10.
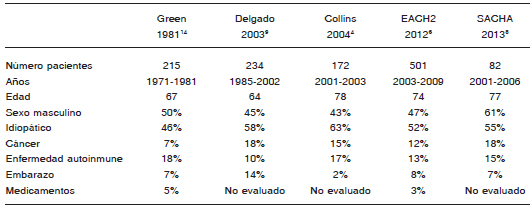
Tabla 1. Características de los pacientes incluidos en los registros internacionales de hemofilia adquirida
Se han determinado algunos marcadores de mal pronóstico, como la permanencia del inhibidor a pesar del tratamiento inmunosupresor, edad mayor a 65 años, hemoglobina menor a 8 g/dl al diagnóstico, título del inhibidor > 15 U Bethesda y tipo de enfermedad asociada (peor pronóstico en hemofilia adquirida asociada al cáncer, luego a enfermedad inmune y rara vez fatal en la asociada a embarazo)5, 11, 13.
Si bien los pacientes con hemofilia congénita también pueden desarrollan inhibidor del FVIII, su comportamiento clínico, tipo de anticuerpo, cinética y hasta el profesional que se enfrenta con el paciente por primera vez es muy diferente al caso de la HA2. A diferencia del aloanticuerpo que aparece en la hemofilia A congénita tras la exposición a FVIII sustitutivo, el autoanticuerpo de la HA tiene una cinética de eliminación diferente, que muestra una curva no lineal, o de segundo orden, especialmente cuando la potencia del anticuerpo es baja. Esto determina que, aun con títulos bajos del inhibidor el efecto neutralizador sigue presente, y también el riesgo de sangrado. En la HA es habitual encontrar niveles residuales de FVIII de 3 a 5%, y en ocasiones hasta 10% que, sin embargo, no evitan la ocurrencia de sangrado grave1, 2.
Diagnóstico de laboratorio en la hemofilia adquirida
Un paciente con autoanticuerpos contra FVIII tiene típicamente un aPTT prolongado con un TP normal y un tiempo de trombina (TT) normal12. Un aPTT prolongado aislado podría deberse a la presencia de un inhibidor lúpico (IL), un inhibidor especifico de un factor, heparina o a un déficit de factores de la coagulación. Es decir que para hacer el diagnóstico de inhibidor de FVIII deben realizarse ensayos para descartar estas alternativas. (Fig. 2). El inhibidor lúpico mimetiza en parte los hallazgos que se ven en la hemofilia adquirida (aPTT prolongado que no corrige en la mezcla con plasma normal) por lo que se recomienda excluirlo realizando pruebas específicas. La presencia de heparina se descarta realizando el TT, que se encuentra alterado. Ante un aPTT prolongado debe realizarse el estudio de mezclas para poder discriminar entre inhibidores y falta o déficit de algún factor puntual de la cascada de coagulación. En el caso del inhibidor específico de FVIII, el aPTT de la mezcla se prolongará aún más si se lo incuba (por ejemplo, una hora a 37 °C) porque el inhibidor de FVIII es tiempo y temperatura dependiente. El paciente con hemofilia adquirida tendrá además un nivel plasmático de FVIII disminuido o ausente. Una vez detectada la presencia de un inhibidor específico de FVIII, este se debe cuantificar con el método Bethesda14, 15 que mide la potencia del anticuerpo. La titulación por este método puede subestimar los títulos en caso de HA porque los autoanticuerpos tienen una cinética no lineal (denominada de tipo II) entre potencia del inhibidor y actividad del FVIII.

Fig. 2. Algoritmo para el diagnóstico de la hemofilia adquirida.
Tratamiento
El objetivo del tratamiento de la HA es detener el sangrado con agentes hemostáticos, y erradicar el inhibidor. También debemos considerar ciertas medidas generales, como minimizar los procedimientos invasivos (aun aquellos considerados menores, como una venopuntura) y llevar a cabo el tratamiento de la enfermedad concomitante, en el caso de identificar alguna. Debemos tener en cuenta que el riesgo de hemorragia grave permanece mientras el inhibidor esté presente, y este riesgo no es predecible ni por la potencia del inhibidor, por el nivel de FVIII residual, y tampoco cuando la forma de presentación clínica inicial es “no grave”3, 15.
En algunas situaciones, tratar la enfermedad asociada es tan importante como tratar al inhibidor mismo. Así, por ejemplo en las enfermedades autoinmunes o en los tumores malignos, se debe priorizar el tratamiento conjunto de ambas enfermedades para obtener la erradicación del inhibidor y detener el sangrado10.
Control de hemostasia
El control de los episodios de sangrado mayor en la HA puede ser difícil y debe ser supervisado por un experto, ya que el tratamiento es muy específico e individualizado para cada paciente. Generalmente la mayoría de los sangrados subcutáneos son benignos, y los sangrados mucosos o musculares suelen ser más severos. Luego de controlada la hemorragia, se debe continuar con la administración de los agentes hemostáticos durante cierto tiempo a fin de prevenir un nuevo sangrado, especialmente en casos de hemorragia en el SNC, retroperitoneo y tejido muscular16-18.
En esta enfermedad no existe correlación entre los niveles de FVIII, el título del inhibidor y el fenotipo hemorrágico. Por lo tanto, ante un episodio de sangrado mayor, el título bajo del inhibidor o el nivel residual de FVIII no deben influir en la decisión del médico sobre el tipo de tratamiento para administrar al paciente15. Las imágenes muchas veces no son útiles por ser tardías. En cambio, sí son útiles para seguir la evolución y para medir la eficacia del tratamiento hemostático16.
Algunos sangrados requieren la administración urgente de soporte hemostático, como en el caso de los localizados en cabeza y cuello, SNC y retroperitoneo, entre otros. También el sangrado que ocurre en múltiples localizaciones en forma simultánea7. En cambio, otros sangrados que no conllevan un riesgo inminente permiten una conducta más conservadora. En ocasiones la hemorragia de las mucosas se beneficia con el tratamiento concomitante con antifibrinolíticos, y en algunos casos con adhesivo tópico de fibrina.
Como fuera mencionado previamente, incluso procedimientos menores como inyecciones intramusculares, exponen al paciente a un sangrado grave, y deben ser evitados.
Un apartado especial merece el caso de la paciente con sangrado post parto sometida a una histerectomía en un centro no preparado para controlar al inhibidor, o el caso de un paciente con síndrome compartimental, en el que se realiza una cirugía de debridamiento del miembro sin el manejo hemostático apropiado.
Aproximadamente un tercio de los pacientes con HA tendrá un segundo evento hemorrágico, y hasta 10% tres o más episodios de sangrado mayor6, por lo que es importante resaltar que el tratamiento no culmina tras el control del sangrado inicial.
Agentes de puenteo (productos que sortean el efecto del inhibidor)
Están indicados siempre en presencia de un sangrado grave, ya sean inhibidores de título alto (mayor a 5 UB) o no. Tanto el factor VII activado recombinante (rFVIIa) como el concentrado de factores protrombínicos activados (CFPa) son agentes de primera línea18, 19. Ambos han demostrado ser eficaces y no existen publicaciones que sugieran la superioridad de alguno de ellos. Para valorar la respuesta, se deben utilizar parámetros indirectos como el cese del dolor por compresión, la estabilización del hematocrito o imágenes que demuestren ausencia de progresión o regresión del hematoma.
El rFVIIa es indicado en una dosis de 90 μg/kg cada 2-6 horas con una respuesta superior al 75%, siendo mayor la efectividad cuando se usa como primera línea de tratamiento17, 19-24. Otro agente de puenteo que se puede utilizar es el CFPa15, 18. La dosis recomendada es 50-100 U/kg cada 8-12 horas (dosis máxima diaria 200 U/kg), con una respuesta similar al anterior24.
Ambos agentes hemostáticos pueden asociarse con eventos trombóticos arteriales y venosos, especialmente en los pacientes añosos cuando se suman otros factores de riesgo (neoplasia, reposo, sepsis o hematoma compresivo)21-25. También debe considerarse la presencia de factores de riesgo cardiovascular que, en esta población añosa, pueden ser determinantes de eventos trombóticos.
Si bien no hay estudios que comparen directamente a los 2 tratamientos antes mencionados (Tabla 2), algunos de los argumentos por los que se podría preferir el rFVIIa serían su mayor seguridad al ser recombinante, disponibilidad inmediata en muchos servicios de cuidados intensivos, menor tiempo de preparación e infusión, estar aprobado para esta indicación por las autoridades regulatorias de salud en Europa y EE.UU. y la mayor experiencia en registros recientes como el EACH2 (European Acquired Haemophilia Registry). Estas ventajas deben balancearse con un costo mayor por unidad en el tratamiento con rFVIIa, si bien no existen trabajos prospectivos de costo-efectividad.
Tabla 2. Características de los agentes de puenteo

Monitoreo del tratamiento
Actualmente no contamos con una técnica de monitoreo de laboratorio validada para definir si se ha corregido la hemostasia en un paciente con HA. Por otro lado, los excelentes resultados obtenidos para el control de inhibidores en hemofilia congénita no pueden extrapolarse a la hemofilia adquirida debido a que la cinética de los inhibidores y los fenotipos hemorrágicos son diferentes.
Duración del tratamiento con agentes de puenteo
Debe mantenerse el tratamiento hasta el cese del sangrado según juicio clínico y criterios indirectos: hematocrito estable, que se controle el dolor, que el tamaño y tensión del hematoma disminuyan, un nivel de FVIII > 30%, o título del inhibidor < 1UB. Usualmente alcanza con menos de 72 horas aunque esto podría resultar insuficiente en ciertos sangrados graves como SNC, retroperitoneo y piso de boca.
Ante el fracaso con uno de estos agentes, se puede considerar utilizar el otro (esperar 4 horas luego de rFVIIa y 6 h luego de CFPa18).
Han sido descriptos para uso en forma excepcional la utilización de tratamiento denominados “de salvataje” en casos extremos o en pacientes que no responden a la terapia habitual, basados en la utilización de plasmaféresis con columnas de inmunoadsorción para retirar los autoanticuerpos, y luego reponer FVIII. En la Argentina no contamos con experiencia en este tipo de tratamiento, y debería considerarse por el momento como experimental16.
Terapia de reemplazo
En cuanto al uso de otros agentes hemostáticos, no se recomienda el concentrado de FVIII humano. De la misma manera, el uso de desmopresina debe limitarse al tratamiento de hemorragias leves o en el contexto de procedimientos invasivos menores. Especialmente en pacientes con un título de inhibidor menor a 3-5 UB, nivel de FVIII residual mayor al 5%, o cuando no se cuenta con agentes de puenteo como primera línea de tratamiento5, 15-17.
El FVIII porcino ha sido aprobado recientemente por la FDA. Esta molécula ha mostrado eficacia, pero no es un producto al que, por el momento, se tenga acceso en nuestro país. Dado la escasa reactividad cruzada con el autoanticuerpo humano y que el FVIII porcino es de origen recombinante, podría tratarse de una alternativa interesante desde el punto de vista del costo-efectividad, y con un menor potencial trombótico3.
Los concentrados de factores protrombínicos “no activados” (CFP) han sido utilizados con éxito en algunos pacientes con HA cuando no se contaba con agentes de derivación, por lo que podrían considerarse ante una emergencia15.
El futuro promete la llegada de mutantes de FIX, zimógenos inactivos capaces de activar FIX independientemente del FVIII y con una vida media más larga que los agentes de puenteo actualmente en uso26.
Erradicar el inhibidor
El objetivo final del tratamiento de la HA es lograr la erradicación completa del inhibidor, lo que se consigue cuando el título del anticuerpo es menor a 0.6 UB, y los niveles de FVIII son mayores al 50%16.
En la mayoría de los casos se debe tratar de instituir la terapia inmunosupresora apenas se establezca el diagnóstico de HA, y aun si el paciente se encuentra asintomático. Cuando el inhibidor no es erradicado, puede ocurrir un episodio de sangrado grave, incluso muchos meses después del comienzo de la enfermedad9. Ni siquiera la forma de presentación inicial como sangrado leve, es lo suficientemente segura como para predecir a largo plazo que no ocurrirá un episodio de sangrado con riesgo de vida.
Sin embargo, hasta un tercio de los casos de HA el inhibidor puede desaparecer espontáneamente en un lapso variable, generalmente menor a 6 meses. Esto es particularmente evidente en niños, aquellos asociados a fármacos y en inhibidores posparto. En estos casos, en tanto no haya manifestaciones de sangrado, la terapia de erradicación puede posponerse 4 a 8 semanas, a la espera de la desaparición espontánea del inhibidor, en especial si el título del mismo es bajo y el nivel residual de FVIII es > 15%27.
Para eliminar al anticuerpo existen diferentes alternativas basadas en tratamiento con inmunosupresores. Sin embargo, contamos con muy pocos trabajos prospectivos que brinden evidencia adecuada sobre cuál es el mejor esquema para utilizar en esta población con alta morbilidad15, 23. En una revisión sistemática, en el 75% de los pacientes tratados con inmunosupresores se logró erradicar el inhibidor9. La mejor combinación, sugerida como primera línea de tratamiento, fue prednisona 1 mg/kg/día asociado a ciclofosfamida 1.5-2 mg/kg/día por 4-6 semanas. Varios trabajos mostraron la alta efectividad y mayor rapidez de respuesta con este esquema. Sin embargo, la mortalidad fue similar a la de los pacientes tratados únicamente con corticoides, probablemente debido a una mayor toxicidad asociada al uso de ciclofosfamida en pacientes añosos. Por lo tanto, la sugerencia actual es que este esquema combinado debe ser utilizado con mucha precaución, ajustando la dosis de ciclofosfamida a 50 mg/día como dosis máxima por 3-4 semanas en pacientes frágiles o con deterioro del estado general18. La asociación de prednisona y ciclofosfamida está especialmente indicada en aquellos pacientes en los que se halla título de inhibidor > 100 UB y con niveles de FVIII menor a 1%, o en aquellos secundarios a enfermedades autoinmunes.
En la HA asociada al embarazo así como en mujeres en edad reproductiva, no se debe utilizar ciclofosfamida, dado el riesgo de esterilidad secundaria al uso de este agente citotóxico29. En estos casos se ha empleado con éxito azatioprina.
Con tratamiento inmunosupresor el tiempo medio hasta la eliminación del inhibidor es de 4-6 semanas, pero muchas veces se detecta más temprano una caída en el título del inhibidor, o una recuperación de los niveles de FVIII16.
La evidencia disponible sugiere que el uso de inmunoglobulina intravenosa como monoterapia o combinada con esteroides, no es efectiva en la erradicación del inhibidor en la HA, por lo que no se recomienda su uso27.
Otra forma de tratamiento inmunosupresor que se ha probado en HA es el rituximab, un anticuerpo monoclonal contra el antígeno CD20 del linfocito B responsable de la formación del anticuerpo contra el FVIII. La dosis evaluada en estos casos fue extrapolada de aquella usada en patología onco-hematológica (375 mg/m² por 4 semanas), aunque se han informado casos aislados de éxito con dosis menores (100 mg/m² por 4 semanas). Esta nueva forma de tratamiento se ha utilizado especialmente en pacientes con enfermedad autoinmune asociada. También en un subgrupo de pacientes con HA que muestran activación del linfocito B o sobre expresión de BAFF/Blas28. Por el momento no contamos con estudios prospectivos que comparen en forma directa este anticuerpo monoclonal con la asociación de prednisona-ciclofosfamida y tampoco ha sido autorizado por las autoridades regulatorias su uso en esta indicación. La recomendación actual para el rituximab es como segunda línea de tratamiento junto con corticoides, luego de transcurridas 4-8 semanas sin respuesta a la inmunosupresión inicial. Algunos trabajos recientes lo postulan como primera línea, con un 77% de respuesta en 8 semanas8, 15, 16.
La lista de tratamientos alternativos, utilizados como tercera línea y comunicados como casos aislados en la literatura incluye a la azatioprina, vincristina, 6-mercaptopurina, micofenolato mofetil y ciclosporina.
La recaída del inhibidor puede ocurrir en el 20-40% de los casos, con una mediana de tiempo hasta la recaída de 7.5 meses desde el cese de la terapia inmunosupresora. En la mitad de los casos responderá al mismo esquema inicial, y en el 25% se hará corticoide dependiente. Por esto es que luego de erradicado el anticuerpo se debe controlar a los pacientes al menos por dos años con estudios de laboratorio. Se debe monitorear el aPTT, nivel de FVIII y eventualmente determinar el título del inhibidor. Este control debe ser semanal en las primeras 6 semanas, mensual en los 6 primeros meses, cada 2-3 meses en el primer año y cada 6 meses luego del año15.
El inhibidor adquirido post parto desaparece espontáneamente en 60 - 100% de los casos en una media de 30 meses. Sin embargo este tiempo puede resultar demasiado largo para adoptar “siempre” una conducta expectante. La complicación más frecuente es el sangrado posparto. Generalmente no vuelve a ocurrir en el siguiente embarazo pero, y en especial si el inhibidor no desapareció, puede tener una respuesta anamnésica29. En un 15% de los casos, el inhibidor puede aparecer durante el embarazo, dando lugar a una complicación menos frecuente pero más grave: el sangrado uterino en el parto.
En conclusión, el manejo de la hemofilia adquirida tiene trascendencia para el hematólogo general y para el médico clínico e intensivista, porque generalmente se presenta como una hemorragia incontrolable en un paciente sin historia de coagulopatía. Los procedimientos más simples en un paciente con sangrado grave, como colocar una vía central o una sonda vesical, pueden desencadenar complicaciones mayores debido a nuevos sangrados. Se requiere de un diagnóstico rápido, para lo cual es imprescindible pensar en esta enfermedad. El basal de hemostasia mostrará un tiempo de protrombina y de trombina normal y un aPTT siempre prolongado, que no corrige con las pruebas de mezcla con plasma normal y que se prolonga marcadamente cuando la mezcla se incuba 1-2 horas a 37 °C. Las medidas de hemostasia habitual como transfusiones de plasma fresco congelado, crioprecipitados y hasta concentrados de FVIII humano son ineficaces, perpetuando el sangrado y retrasando el inicio del tratamiento adecuado. Debemos utilizar agentes de puenteo, como el rFVIIa y el CFPa, los cuales, debido a su uso infrecuente y alto costo, no suelen estar fácilmente disponibles en hospitales generales. Al mismo tiempo, el tratamiento con fármacos inmunosupresores debe ser temprano y adecuado. Se requiere un uso prudente de los agentes citotóxicos en esta población añosa, frágil, o de mujeres en edad fértil. La combinación de prednisona y ciclofosfamida es la primera elección de tratamiento en la mayoría de los pacientes, ajustando la dosis y duración de la ciclofosfamida en los casos de mayor riesgo de mielotoxicidad. En pacientes que no puedan recibir ciclofosfamida o ante la falta de respuesta a la primera línea, se puede usar rituximab, aunque la tendencia actual es a adelantar cada vez más el uso de este anticuerpo monoclonal.
Conflicto de intereses: Ninguno para declarar
1. Baudo F, de Cataldo F. Acquired hemophilia: a critical bleeding syndrome. Haematologica 2004; 89: 96-100. [ Links ]
2. Webert KE. Acquired hemophilia A. Semin Thromb Hemost 2012; 38: 735-41. [ Links ]
3. Coppola A, Favaloro EJ, Tufano A, Di Minno MN, Cerbone AM, Franchini M. Acquired inhibitors of coagulation factors: part I-acquired hemophilia A. Semin Thromb Hemos 2012; 38: 433-46. [ Links ]
4. Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007; 109: 1870-7.
5. Franchini M, Lippi G. How I treat acquired factor VIII inhibitors. Blood 2008 15; 112: 250-5. [ Links ]
6. Knoebl P, Marco P, Baudo F, et al. EACH2 registry contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10: 622-31. [ Links ]
7. Bitting RL, Bent S, Li Y, Kohlwes J. The prognosis and treatment of acquired hemophilia: a systematic review and meta-analysis. Blood Coagul Fibrinolysis 2009; 20: 517-23. [ Links ]
8. Borg JY, Guillet B, Le Cam-Duchez V, Goudemand J, Lévesque H; SACHA Study Group. Outcome of acquired haemophilia in France: the prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hémophilie Acquise) registry. Haemophilia 2013; 19: 564-70.
9. Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol 2003; 121: 21-35. [ Links ]
10. Alzate M, Meschengieser S, Blanco A, Grosso S, Lazzari M, Sanchez-Luceros A. Acquired Hemophilia A. Experience of a single center. Blood 2013; 122: 4781. [ Links ]
11. Collins PW. Treatment of acquired haemophilia. J Thromb Haemost 2007; 5: 893-900. [ Links ]
12. Franchini M. Postpartum acquired factor VIII inhibitors. Am J Hematol 2006; 81: 768-73. [ Links ]
13. Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to Factor VIII. Thromb Haemost 1981; 45: 200-3. [ Links ]
14. Verbruggen B. Diagnosis and quantification of FVIII inhibitors. Haemophilia 2010; 16: 20-24. [ Links ]
15. Verbruggen B, Novaková I, van Heerde W. Detecting and quantifying functional and inhibitor in hemostasis. In: S Kitchen, J Olson and E Preston. Quality in Laboratory Hemostasis and Thrombosis. Blackwell Publishing, p 198-207. [ Links ]
16. Sborov DW, Rodgers GM. How I manage patients with acquired haemophilia A. Br J Haematol. 2013; 161: 157-65. [ Links ]
17. Baudo F, Collins P, Huth-Kühne A, et al. EACH2 registry contributors. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry. Blood. 2012; 120: 39-46. [ Links ]
18. Zeitler H, Ulrich-Merzenich G, Goldmann G, Vidovic N, Brackmann HH, Oldenburg J. The relevance of the bleeding severity in the treatment of acquired haemophilia - an update of a single-center experience with 67 patients. Haemophilia 2010; 16: 95-101. [ Links ]
19. Abshire T, Kenet G. Recombinant factor VIIa: review of efficacy, dosing regimens and safety in patients with congenital and acquired factor VIII or IX inhibitors. J Thromb Haemost 2004; 2: 899-909. [ Links ]
20. Croom KF, McCormack PL. Recombinant factor VIIa (eptacog alfa): a review of its use in congenital hemophilia with inhibitors, acquired hemophilia, and other congenital bleeding disorders. BioDrugs 2008; 22: 121-36. [ Links ]
21. Franchini M. Recombinant factor VIIa: a review on its clinical use. Int J Hematol 2006; 83: 126-38. [ Links ]
22. Sumner MJ, Geldziler BD, Seremetis S. Treatment of acquired haemophilia with recombinant activated FVII: a critical appraisal. Haemophilia 2007; 13: 451-61. [ Links ]
23. Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: a multicentre study. Thromb Haemost 1997; 78: 1463-7. [ Links ]
24. Aledort LM. Factor VIII inhibitor bypassing activity (FEIBA) – addressing safety issues. Haemophilia 2008; 14: 39-43.
25. Aledort LM. Comparative thrombotic event incidence after infusion of recombinant factor VIII inhibitor bypass activity. J Thromb Haemost 2004: 2: 1700-8 [ Links ]
26. Quade-Lyssy P, Abriss D, Milanov P et al. Next generation FIX muteins with FVIII-independent activity for alternative treatment of hemophilia A. J Thromb Haemost 2014; 12: 1861-73. [ Links ]
27. Collins P, Baudo F, Knoebl P et al. EACH2 registry collaborators. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood 2012; 120: 47-55. [ Links ]
28. Sakurai Y, Takeda T. Acquired hemophilia: a frequently overlooked acquired hemorrhagic disorder: J Immunol Res 2014; 2014: 320674. [ Links ]
29. Tengborn L, Baudo F, Huth-Kuhne A et al. EACH2 registry contributors. Pregnancy-associated acquired haemophilia A: results from European Acquired Haemophilia (EACH2) registry. BJOG 2012; 119: 1529-37. [ Links ]

