










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) vol.76 no.1 Ciudad Autónoma de Buenos Aires feb. 2016
CASUÍSTICA
Síndrome de Davidenkow. Una neuropatía periférica rara
Leonardo Halley Carvalho Pimentel1, Aline Lariessy Campos Paiva2, José Carlos Esteves Veiga2, Guilherme Brasileiro De Aguiar2
1Departamento de Neurologia, Faculdade de Ciências Médicas da Universidade Estadual do Piauí (UESPI), Teresina, Piauí,
2Departamento de Cirurgia, Faculdade de Ciências Médicas da Santa Casa de São Paulo, São Paulo, Brasil
Dirección postal: Guilherme Brasileiro de Aguiar, División de Neurocirugía, Departamento de Cirugía, Faculdade de Ciências Médicas da Santa Casa de São Paul, Rua Cesário Motta Jr., 112, Vila Buarque, 01221-900 São Paulo (SP), Brasil
e-mail: guilhermebraguiar@yahoo.com.br
Recibido: 1-VII-2015
Aceptado: 10-XI-2015
Resumen
En 1939 Davidenkow describió un tipo de atrofia diferente y rara con un patrón predominante en distribución escápulo-peroneal. Algunos investigadores caracterizaron el síndrome como una variante de la enfermedad de Charcot-Marie-Tooth; sin embargo, Davidenkow percibió que las manifestaciones clínicas y de laboratorio no corroboraban exactamente esta hipótesis. Describimos el caso de una mujer de 39 años, con cuadro clínico semejante al síndrome descrito por Davidenkow, presentando atrofia escápulo-peroneal. Sus primeros síntomas comenzaron cuando tenía 24 años, inicialmente con debilidad motora proximal en los miembros superiores. No tenía historia familiar de miopatía o neuropatía y se excluyeron otros síndromes que se podrían incluir entre los diagnósticos diferenciales mediante la realización de pruebas de mutación genética, además del examen físico y electromiografía. El amplio espectro de enfermedades neuromusculares a veces dificulta su diagnóstico y debe ser siempre considerado en el diagnóstico diferencial.
Palabras clave: Síndrome de Davidenkow; Síndrome escápulo-peroneal; Amiotrofia escápulo-peroneal; Atrofia muscular proximal; Atrofia neurogénica escápulo-peroneal.
Abstract
Davidenkow syndrome. A rare peripheral neuropathy.
A different and rare type of atrophy with a predominant pattern in scapulo-peroneal distribution was described by Davidenkow in 1939. The syndrome was characterized by some researchers as a variant of Charcot-Marie-Tooth disease, however Davidenkow noticed that clinical and laboratorial manifestations did not corroborate exactly with this hypothesis. We describe a case of a female patient, 39 years-old, clinical picture similar to the syndrome described by Davidenkow, presenting scapulo-peroneal atrophy. Her first symptoms had appeared when she was 24, initially with proximal motor weakness in the upper limbs. This patient did not have family history of myopathy or neuropathy. Several tests were performed to exclude other syndromes that could be included in the differential diagnosis, by testing gene mutation, in addition to the physical examination and electromyography. The large spectrum of neuromuscular diseases makes difficult the diagnosis of Davidenkow's syndrome which always should be considered in the differential diagnosis.
Key words: Davidenkow syndrome; Scapuloperoneal syndrome; Scapulo-peroneal amyotrophy; Proximal muscle atrophy; Neurogenic scapuloperoneal atrophy.
Davidenkow definió un tipo de atrofia muscular con patrón predominante escapuloperoneal, semejante a la enfermedad de Charcot-Marie-Tooth (CMT)1. Sin embargo, las manifestaciones clínicas y de laboratorio del paciente no correspondían a esta enfermedad, así que se designó como síndrome de Davidenkow (SD)1. En su primera descripción, Davidenkow solamente informó estos nuevos hallazgos y, después, pocos investigadores han usado dicho término para denominar esta enfermedad neuromuscular que afecta predominantemente los músculos escapulares y peroneos, es más frecuente en mujeres y puede o no presentar un patrón familiar.
Se presenta el caso de una paciente cuyas manifestaciones clínicas, hallazgos neurológicos, electrofisiológicos e histológicos son compatibles con SD. Debido a la escasez de informes debidamente investigados y diagnosticados con SD comprobado, también se realizó una breve revisión de la literatura sobre enfermedades neuromusculares similares, discutiendo sus características principales.
Caso clínico
Una mujer de 39 años consultó al Centro de Rehabilitación Integrada en febrero de 2010 por debilidad muscular progresiva y asimétrica en los cuatro miembros, predominantemente en la extremidad superior derecha, que comenzó 15 años atrás. Durante ese período, no tuvo diagnóstico de certeza y recibió tratamiento para el dolor y fisioterapia. Dos años antes padeció atrofia muscular progresiva y paralela de la cintura escapular, con reciente empeoramiento. Anteriormente, se hallaba saludable, no consumía alcohol, no fumaba ni tenía hábitos perniciosos. En la historia familiar (abuelos, padres, hermanos o hijos) no se encontraron cuadros de miopatía, neuropatía u otras afecciones, tales como diabetes, enfermedades autoinmunes o de la columna vertebral. En el examen neurológico se observó marcha laboriosa, especialmente dificultad en la marcha calcánea y tendencia a la rotación externa del miembro derecho inferior. Los hombros, pies (Fig.1) y manos presentaban grave atrofia y se percibió reducción de la fuerza muscular en los miembros superiores -grado III del Consejo de Investigación Médica-, principalmente en el deltoides, tríceps, bíceps y flexores de la muñeca, con predominio en el lado derecho. Todos los grupos musculares del lado izquierdo mostraron fuerza grado IV. El tono muscular y los reflejos de los tendones profundos eran normales y simétricos. Las coordinaciones axial y apendicular estaban preservadas.
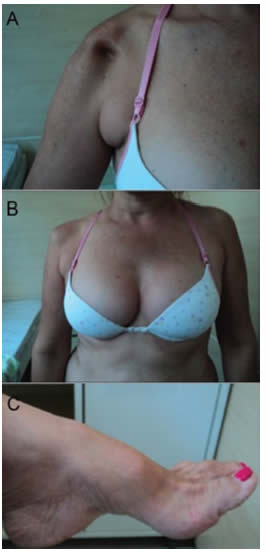
Fig. 1. Alteraciones morfológicas halladas en el examen físico. A: Atrofia de la cintura escapular derecha. B: Asimetría y atrofia muscular del miembro superior derecho. C: Atrofia muscular en el pie izquierdo (pie cavo)
No se observó el engrosamiento de los nervios periféricos. Se percibió aumento de la sensibilidad dolorosa en el hemicuerpo derecho, disminución de sensibilidad de vibración en los miembros inferiores y pérdida de la noción de posición del arco escarzano de los dedos de los pies, con predominio derecho. La sensibilidad táctil y térmica estaban preservadas y no hubo alteraciones en el examen de los nervios cranianos.
Se realizó una electroneuromiografía (ENMG) que reveló falta de excitabilidad del potencial de acción motora en segmentos distales, asociada a la reducción de la extensión de potencial de acción motora en los nervios mediano derecho, peroneo izquierdo y tibial; no se obtuvo la medición de velocidad de conducción sensitiva en los nervios examinados -mediano, ulnar, radial y surales derechos e izquierdos-. Este estudio sugiere polineuropatía motora y sensorial, predominantemente motora con un patrón axónico con señales de evolución crónica de reinervación. Los niveles plasmáticos de enzimas musculares como la creatinfosfoquinasa (valor de referencia < 130 UI/ml) y la aldolasa (valor de referencia < 7.0) fueron normales (46 y 2.3, respectivamente).
Considerándose la situación clínica de la polineuropatía, se realizó la biopsia del bíceps derecho y una evaluación histoquímica complementaria para la ATPasa de pH 4.6 9.4 r, beta-NADH-tetrazolio, esterasa no específica, fosfatasa alcalina, Oil Red O, tricrómico de Gomori, hematoxilina/eosina y cristal violeta con secciones de 8 micras de espesor usando un criostato a -25 °C, que mostró fibras atróficas con señales de denervación, permitiendo el diagnóstico de una neuropatía. Se realizó una evaluación molecular para CMT, buscando la deleción o duplicación del gen PMP22 mediante la técnica multiplex ligation-dependent probe amplification, que resultó normal.
Se diagnosticó síndrome de Davidenkow. El tratamiento con antidepresivos tricíclicos (amitriptilina 50 mg diarios) redujo sustancialmente la debilidad muscular y las alteraciones sensitivas después de 3 meses de uso.
Discusión
Las neuropatías periféricas son enfermedades relativamente comunes, pero el diagnóstico diferencial entre sus múltiples tipos resulta difícil y laborioso. La manifestación clínica cardinal del SD es la debilidad escápulo-peroneal2. Sin embargo, esta debilidad puede ser encontrada en otras neuropatías como la CMT, miopatía de Miyoshi, enfermedad de Dejerine-Sottas y otras3, que tienen en común una disminución de velocidad de conducción nerviosa en el ENMG2.
Hay divergencia entre su nomenclatura y clasificación. Davidenkow sostuvo inicialmente que el síndrome era un tipo de CMT, pero tras algunos años concluyó que, si bien es una entidad independiente, se asemeja más a la enfermedad de Dejerine-Sottas4.
La CMT es una neuropatía hereditaria motora sensitiva que presenta diversos tipos, el más común es el tipo 1A. Se caracteriza por debilidad, atrofia de los músculos distales y pérdida de sensibilidad que evoluciona muy lentamente y está asociada a pie cavo5; generalmente comienza en la niñez6. A pesar de tener un rango de transmisión genética amplio (autosómica dominante, autosómica recesiva o vinculada al cromosoma X), en la mayoría de los casos ocurre debido a una duplicación en el cromosoma 17p, gen PMP227.
Otra neuropatía que se debe considerar en el diagnóstico diferencial es la enfermedad de Dejerine-Sottas, también conocida como neuropatía intersticial hipertrófica8. Esta es aún más rara que la CMT y aparece en la niñez como una debilidad progresiva, pérdida de sensibilidad en los miembros inferiores, frecuentemente evolucionando a la necesidad del uso de silla de ruedas8. Generalmente la velocidad de conducción nerviosa9 es menor de 12 m/s (en este caso la velocidad variaba entre 48 y 54 m/s entre los diversos nervios periféricos analizados). La debilidad muscular progresa más rápidamente que la de CMT. Generalmente ambas neuropatías afectan al nervio facial3.
La distrofia de Miyoshi es una condición rara, que se describió inicialmente en Japón10. Presenta una herencia autosómica recesiva, ocurre principalmente en familias que tienen matrimonios consanguíneos10. Se caracteriza por comenzar con debilidad y atrofia muscular y, a veces, exclusivamente en los músculos posteriores de los miembros inferiores, con atrofia grave del gastrocnemio y tiene un patrón de evolución lento. Recientemente se registró que, relacionada a esta distrofia, hay una alteración genética del cromosoma 2p12-1411. Esta enfermedad evoluciona con altos niveles de creatinfosfoquinasa y una notable disminución de los niveles de disferlina, que es uno de los componentes de la membrana de la fibra muscular11.
La enferma que describimos no presentaba ningún patrón de herencia genética, así que es probable que esta sea una enfermedad esporádica. Asimismo, es poco probable la presencia de las otras enfermedades mencionadas anteriormente. Al igual que el ENMG, la evaluación histopatológica no reveló el patrón de miopatía. Corroborando la descripción inicial realizada por Davidenkow, esta enfermedad tiene sus manifestaciones clínicas durante la vida adulta1. Los análisis electrofisiológicos e histológicos también confirmaron la hipótesis que la evolución y los hallazgos del examen físico no estaban de acuerdo con otras neuropatías comunes. Además, en este caso no se requirió la biopsia de los nervios para establecer el diagnóstico de síndrome de Davidenkow, ya que la biopsia muscular y el estudio genético fueron suficientes para el diagnóstico.
El tratamiento del SD busca la reducción de síntomas. Su pronóstico y evolución es variable, es una enfermedad neurodegenerativa para la cual no hay una terapia efectiva que permita revertir el déficit anterior. Los medicamentos que más se evaluaron para el tratamiento sintomático son los antidepresivos tricíclicos, especialmente la amitriptilina2, 9, asociada a la rehabilitación física. En este caso, se utilizó un antidepresivo tricíclico y la rehabilitación, con mejora significativa de los síntomas.
El amplio espectro de enfermedades musculares con una participación variable de diversos grupos de músculos puede dificultar el diagnóstico etiológico preciso. La ausencia de herencia, asociada a los síntomas de la edad adulta que son compatibles con la definición de Davidenkow, y el análisis histopatológico, son factores determinantes para el diagnóstico de SD.
Conflicto de intereses: Ninguno para declarar
1. Schwartz MS, Swash M. Scapuloperoneal atrophy with sensory involvement: Davidenkow's syndrome. J Neurol Neurosurg Psychiatry 1975; 38: 1063-7. [ Links ]
2. Ricker K, Mertens HG, Schimirig Schimrigk K. The neurogenic scapulo-peroneal syndrome. Eur Neurol 1968; 1: 257-74. [ Links ]
3. Amici SA, Dunn WA Jr, Murphy AJ, et al. Peripheral myelin protein 22 is in complex with alpha6beta4 integrin, and its absence alters the Schwann cell basal lamina. J Neurosci 2006; 26: 1179-89. [ Links ]
4. Kazakov V. What is Davidenkov's scapuloperoneal amyotrophy: is it a myopathic entity or a neurogenic syndrome? What was Davidenkov's opinion concerning this knotty problem? Neuromuscul Disord 2003; 13: 91-2. [ Links ]
5. Maranho DAC, Volpon JB. Pé cavo adquirido na doença de Charcot-Marie-Tooth. Rev Bras Ortop 2009; 44: 479-86. [ Links ]
6. Berciano J, Sevilla T, Casasnovas C, et al. Guía diagnóstica en el paciente con enfermedad de Charcot-Marie-Tooth. Neurología 2012; 27: 169-78. [ Links ]
7. Verhamme C, van Schaik IN, Koelman JH, de Haan RJ, de Visser M. The natural history of Charcot-Marie-Tooth type 1A in adults: a 5-year follow-up study. Brain 2009; 132: 3252-62. [ Links ]
8. Marinho JL, Alonso Nieto JL, Calore EE. Dejerine-Sottas disease: a case report. Sao Paulo Med J 2003; 121: 207-9. [ Links ]
9. Gabreëls-Festen A. Dejerine-Sottas syndrome grown to maturity: overview of genetic and morphological heterogeneity and follow-up of 25 patients. J Anat 2002; 200: 341-56. [ Links ]
10. Soares CN, de Freitas MR, Nascimento OJ, da Silva LF, de Freitas AR, Werneck LC. Myopathy of distal lower limbs: the clinical variant of Miyoshi. Arq Neuropsiquiatr 2003; 61: 946-9. [ Links ]
11. Bejaoui K, Hirabayashi K, Hentati F, et al. Linkage of Miyoshi myopathy (distal autosomal recessive muscular dystrophy) locus to chromosome 2p12-14. Neurology 1995; 45: 768-72. [ Links ]

