










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina (Buenos Aires)
versión impresa ISSN 0025-7680versión On-line ISSN 1669-9106
Medicina (B. Aires) vol.78 no.3 Ciudad Autónoma de Buenos Aires jun. 2018
CASUÍSTICA
Panhipopituitarismo y escotoma perimacular como presentación de enfermedad relacionada a IgG4
Gabriela A. Sosa1, Patricia Fainstein-Day2, Silvia Christiansen3, Pablo Ajler4, Claudio Yampolsky4
1 Servicio de Endocrinología, Hospital Provincial de Unquillo, Córdoba,
2 Servicio de Endocrinología, Metabolismo y Medicina Nuclear,
3 Servicio de Patología,
4 Servicio de Neurocirugía, Hospital Italiano de Buenos Aires, Argentina
Dirección postal: Patricia Fainstein-Day, Hospital Italiano de Buenos Aires, Tte. Gral. Juan D. Perón 4190, 1199 Buenos Aires, Argentina
e-mail: patricia.fainstein@hospitalitaliano.org.ar
Recibido: 9-VIII-2017
Aceptado: 31-X-2017
Resumen
La enfermedad relacionada a IgG4 (IgG4-RD) constituye una entidad sistémica recientemente descrita, de causa desconocida. Afecta predominantemente a hombres mayores y presenta características histopatológicas distintivas, como fibrosis estoriforme, flebitis obliterante y denso infiltrado linfoplasmocitario con inmunomarcación para IgG4, pudiendo estar asociada a elevación sérica de dicha inmunoglobulina. Si bien cualquier órgano puede estar afectado, el compromiso de la hipófisis es infrecuente. Describimos el caso de un hombre de 36 años que se presentó con cefaleas, alteración del campo visual, panhipopituitarismo, diabetes insípida y una imagen que mostraba una lesión infiltrativa infundíbulo-panhipofisaria extendida. Arribamos al diagnóstico de IgG4-RD a través de biopsia hipofisaria. La respuesta al tratamiento con dosis inmunosupresoras de corticoides fue exitosa.
Palabras clave: Panhipopituitarismo; Hipofisitis; IgG4; Escotoma.
Abstract
IgG4 related disease presenting as panhypopituitarism and perimacular scotoma.
IgG4-related disease (IgG4-RD) is a recently described systemic entity of unknown origin. It predominantly affects older men and has distinctive histopathologic features as storiform fibrosis, obliterative phlebitis, dense lymphoplasmacytic infiltrate with immunostaining for IgG4, and it may be associated with elevated serum levels of IgG4. Although any organ can be affected, pituitary gland is rarely involved. We describe the case of a 36-year-old man who presented with headaches, impaired vision, panhypopituitarism with diabetes insipidus and an infiltrative lesion mainly of infundibulum and pituitary. We arrived at diagnosis of IgG4-RD by pituitary biopsy. A successful response to treatment with immunosuppressive doses of corticosteroids was achieved.
Key words: Panhypopituitarism; Hypophysitis; IgG4; Scotoma.
La enfermedad relacionada a inmunoglobulina G4 (IgG4-RD, del inglés: IgG4-related disease) fue descrita por primera vez en el año 2001 a partir del estudio de la pancreatitis esclerosante1. Hoy se sabe que muchas entidades consideradas de origen idiopático, como la tiroiditis de Riedel, la fibrosis retroperitoneal, la nefritis tubulointersticial idiopática y el síndrome de Mikulicz, entre otros, pertenecen a esta entidad. Se pueden presentar síntomas compresivos u obstructivos debidos a organomegalia o hipertrofia; o disfunción por infiltración celular y fibrosis2. En los últimos años se ha progresado en el conocimiento de la enfermedad, lo que ha permitido su sospecha y diagnóstico. Esta es la clave para que el tratamiento oportuno se realice antes de la etapa de fibrosis masiva, hasta ahora irreversible. Presentamos el caso de un hombre joven cuya primera manifestación fue alteración grave de la visión.
Caso clínico
Varón, de aparente origen caucásico, de 36 años, derivado al Servicio de Endocrinología del Hospital Italiano de Buenos Aires, luego de haberse efectuado una biopsia hipofisaria debido a masa ocupante selar. Paciente obeso, sin antecedentes de hábitos tóxicos ni consumo de fármacos, refería un cuadro de dos meses de evolución de cefaleas, escotomas y fotopsias en ojo derecho; astenia, poliuria y polidipsia.
El examen físico no fue relevante: índice de masa corporal (IMC) 32, con signos vitales dentro de parámetros normales, ausencia de foco neurológico, sin signos de hipogonadismo y palpación tiroidea normal. La resonancia magnética (RM) de región selar evidenciaba hipófisis agrandada con invasión supraselar y engrosamiento del tallo hipofisario, infiltración del quiasma, nervios ópticos y esfenoides (Fig. 1 A, B, C).
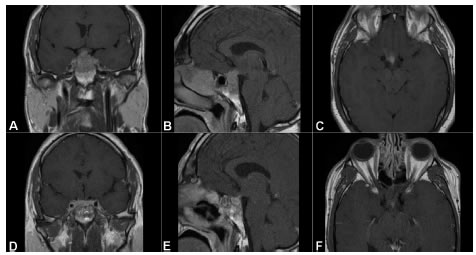
Fig. 1. RM de hipófisis con gadolinio. A) B) C): cortes coronal, sagital y axial, respectivamente. Lesión selar con extensión supraselar e invasión de esfenoides, previo a tratamiento corticoideo. D) E) F): Reducción del tamaño de la lesión tras tres meses de tratamiento
El hemograma, función renal, función hepática e ionograma estaban dentro de parámetros normales. La eritrosedimentación era de 40 mm/h y la enzima convertidora de angiotensina (ECA) de 93 μg/l, VN (valor normal) < 52 μg/l). El laboratorio hormonal informó TSH < 0.01 μUI/ml (VN 0.4 - 4 μUI/ml), T4L 0.6 ng/ml (VN 0.8- 2 ng/dl), ATPO 365 UI/ml (VN < 34 UI/ml), ATG 58 UI/ml (VN <40 UI/ml), cortisol plasmático 8 horas < 1 μg/dl (VN 5- 18 μg/dl), cortisol libre urinario <40 μg/ml (VN < 100 μg /ml), testosterona total <0.2 ng/ml (VN 0.08- 0.5 ng/ml), LH <0.5 mUI/ml (VN 2- 5 mUI/ml), FSH 1.3 ng/ml (VN 1-8 UI/ml), IGF-1 276 ng/ml (VN 64- 284 ng/ml), prolactina 24.5 ng/ml (VN < 20 ng/ml). Estos resultados fueron compatibles con panhipopituitarismo por compromiso de los ejes corticotropo, tirotropo y gonadotropo. Los niveles séricos de IgG total y de IgG4 fueron normales. El campo visual computarizado mostró escotoma perimacular derecho.
Se indicó tratamiento sustitutivo con hidrocortisona y a la semana se inició con levotiroxina. El resultado de la biopsia informó infiltrado linfoplasmocitario y fibrosis estoriforme con flebitis concéntrica, por lo que se realizó citometría de flujo que descartó monoclonalidad. La inmunomarcación fue positiva para IgG4 en más de 10 plasmocitos por campo de gran aumento, confirmando el diagnóstico de IgG4-RD (Figura 2). Una tomografía axial computarizada de tórax, abdomen y pelvis descartó compromiso de otros órganos.
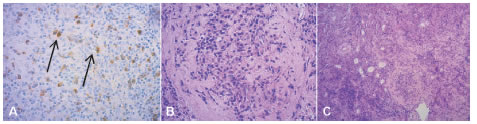
Fig. 2. Biopsia de hipófisis. A) Inmunomarcación positiva para IgG4 en más de 10 plasmocitos por campo de 40X (flechas). B) Infiltrado mononuclear rico en plasmocitos y abundante estroma colágeno (40X). C) Fibrosis estoriforme y flebitis concéntrica (10X)
Se cambió la hidrocortisona por meprednisona, 40 mg por día, durante tres meses, presentando notable mejoría: recuperación de la visión, reversión de la diabetes insípida y reducción del tamaño lesional (Fig. 1D, E, F). Luego del descenso gradual de la dosis hasta 4 mg por día, a los 9 meses de tratamiento continúa estable.
Discusión
Las IgG4-RD constituyen una condición fibroinflamatoria sistémica de reciente conocimiento, que compromete más frecuentemente a hombres en la sexta década de la vida. Su causa es desconocida, aunque un origen alérgico ha sido propuesto dada su frecuente asociación a elevación de IgE, eosinofilia y atopía. Actualmente, el 74% del total de casos han sido comunicados en Japón, con una incidencia de 0.28-1.08 por 100 000 habitantes/año2, 3. Pueden estar afectados varios órganos, en forma sincrónica o metacrónica y con un curso solapado. Arribamos al diagnóstico en base a la clínica y la histopatología, con fibrosis estoriforme, flebitis obliterante e infiltrado linfoplasmocitario con inmunomarcación positiva para IgG4 en plasmocitos. Si bien los niveles séricos de IgG4 fueron aquí normales, se observan aumentados en un 50% de los pacientes4. Como en el caso presentado, la región de la cabeza y cuello es una de las más afectadas por las IgG4-RD, siendo las glándulas submandibulares, órbita y tiroides los sitios más frecuentemente comprometidos5.
Contrariamente, las hipofisitis por IgG4 son de presentación poco habitual, manifestándose con diabetes insípida, hipopituitarismo y/o síntomas compresivos6.
Con respecto a los anticuerpos antiperoxidasa y antitiroglobulina, que se hallaron positivos, fueron interpretados como relacionados a tiroiditis autoinmune eutiroidea, descartándose la tiroiditis de Riedel debido a la ausencia de síntomas cervicales compresivos, palpación tiroidea normal y densidad normal de la glándula observada en la tomografía computada7.
La lesión infiltrativa de la hipófisis y el engrosamiento del tallo puede observarse en otras enfermedades, por ejemplo en las hipofisitis primarias, o secundarias a enfermedades sistémicas como la enfermedad de Wegener, histiocitosis de Langerhans, sarcoidosis y tuberculosis8. En nuestro caso no encontramos signos típicos de las mismas o infiltración de otros tejidos biopsiables; y si bien la enzima convertidora de angiotensina (ECA) se encontraba elevada, se trata de un marcador con poca especificidad9. Por lo tanto, solo la histología de la hipófisis podía permitir el diagnóstico diferencial. Es de notar que ante una posible hipofisitis primaria linfocítica la conducta más recomendable es la observación y sustitución de las insuficiencias endocrinas detectadas, que usualmente son transitorias. La imagen, en esos casos, es simétrica y más limitada a hipófisis y afecta típicamente a mujeres jóvenes durante el embarazo y el puerperio10.
Aunque no lo observamos en nuestro paciente, debemos recordar que muchas enfermedades cursan con niveles plasmáticos elevados de IgG4, como las vasculitis, artritis reumatoidea, enfermedad inflamatoria intestinal, rinosinusitis, anemia perniciosa, entre otras; y que algunas presentan infiltración de plasmocitos con inmunohistoquímica positiva para IgG4 (como las neoplasias pancreato-biliares). En todas ellas faltan las características histológicas típicas de las IgG4-RD. Otro diagnóstico diferencial importante es el linfoma B, para lo cual es de utilidad la citometría de flujo4, 11.
La respuesta a terapia inmunosupresora es buena y los glucocorticoides se utilizan en el tratamiento de primera línea. El rituximab ha mostrado resultados satisfactorios en casos de falta de respuesta a glucocorticoides, contraindicación para dosis inmunosupresoras de los mismos, o recidiva de la enfermedad12.
Finalmente, resta mucho por conocer sobre estas enfermedades, sobre su etiología y la respuesta a largo plazo a inmunosupresores; y dado el compromiso sistémico, un manejo multidisciplinario es indispensable.
Conflicto de intereses: Ninguno para declarar
1. Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344: 732-8. [ Links ]
2. Umehara H, Okazaki K, Masaki K, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD). Mod Rheumatol 2012; 22: 21-30. [ Links ]
3. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012; 366: 539-51. [ Links ]
4. Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012; 25: 1181-92. [ Links ]
5. Mulholland GB, Jeffery CC, Satija P, Côté DW. Immunoglobulin G4-related diseases in the head and neck: a systematic review. J Otolaryngol Head Neck Surg 2015; 44: 24. [ Links ]
6. Shikuma J, Kan K, Ito R, et al. Critical review of IgG4-related hypophysitis. Pituitary 2017; 20: 282-91. [ Links ]
7. Hennessey JV. Riedel’s Thyroiditis: A Clinical Review. J Clin Endocrinol Metab 2011; 96: 3031-41.
8. Faje A. Hypophysitis: evaluation and management. Clin Diabetes Endocrinol 2016; 2: 15. [ Links ]
9. Ungprasert P, Carmona EM, Crowson CS, Matteson EL. Diagnostic utility of angiotensin-converting enzyme in sarcoidosis: a population-based study. Lung 2016; 194: 91-5. [ Links ]
10. Khare S, Jagtap VS, Budyal SR, et al. Primary (autoimmune) hypophysitis: a single centre experience. Pituitary 2015; 18: 16-22. [ Links ]
11. Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet 2015; 385: 1460-71. [ Links ]
12. Kamisawa T, Okazaki K. Diagnosis and treatment of IgG4-related disease. Curr Top Microbiol Immunol 2017; 401: 19-33. [ Links ]

