










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina (Buenos Aires)
Print version ISSN 0025-7680On-line version ISSN 1669-9106
Medicina (B. Aires) vol.78 supl.1 Ciudad Autónoma de Buenos Aires Aug. 2018
SUPLEMENTO
Consenso argentino sobre enfermedad de Pompe de inicio tardío
Alberto Dubrovsky1, Ernesto Fulgenzi2, Eduardo L. De Vito3,4, Fabio Barroso5, Andrés Berardo6, Mariela Bettini7, Daniela Binaghi8, Esteban Calabrese9, Daniel Carlés10, Marcelo Chaves11, Fernando Chloca12, Eugenia Conti13, Jose Corderi14, Federico Di Gennaro15, Nélida Ferradás16, Agustín Jáuregui17, Fabiana Lubieniecki18, Claudio Mazia†19, Marta Medina20, Laura Pirra1, Juan Politei21, Ricardo Reisin22, Alberto L. Rosa23, Marcelo Rugiero7, Valeria Salutto19, Andrea Schenone21, Mario Sussini24, Ana L. Taratuto25
1 Instituto de Neurociencias, Fundación Favaloro, Buenos Aires,
2 Servicio de Neurología, Hospital General de Agudos Dr. Ignacio Pirovano, Buenos Aires,
3 Instituto de Investigaciones Médicas Alfredo Lanari, Universidad de Buenos Aires,
4 Cuidados Respiratorios, Centro del Parque, Buenos Aires,
5 Servicio de Enfermedades Neuromusculares, Instituto de Investigaciones Neurológicas-FLENI, Buenos Aires,
6 Unidad de Neurociencias, Instituto Conci Carpinella, Córdoba,
7 Servicio de Enfermedades Neuromusculares, Hospital Italiano de Buenos Aires,
8 Departamento de Imágenes, Fundación Favaloro, Buenos Aires,
9 Servicio de Enfermedades Neuromusculares, INECO, Rosario, Santa Fe,
10 Servicio de Neumonología, Hospital Perrando, Resistencia, Chaco,
11 Servicio de Enfermedades Neuromusculares, Hospital San Martín, Paraná, Entre Ríos,
12 Servicio de Neurología, Hospital Malvinas Argentinas, Buenos Aires,
13 Servicio de Neurología, Área de Enfermedades Neuromusculares, Hospital de Clínicas José de San Martín, Buenos Aires,
14 Departamento de Kinesiología, Fundación Favaloro, Buenos Aires,
15 Departamento de Medicina Interna, Hospital Alemán, Buenos Aires,
16 International Life Sciences Institute (ILSI), Buenos Aires,
17 Departamento de Neurología, Fundación Favaloro, Buenos Aires,
18 Servicio de Patología, Hospital de Pediatría Prof. Dr. J. P. Garrahan, Buenos Aires,
19 Departamento de Neurología, Instituto de Investigaciones Médicas Alfredo Lanari, Universidad de Buenos Aires,
20 Servicio de Neurología, Hospital Córdoba, Córdoba, Argentina,
21 Laboratorio de Neuroquímica Dr. N. A. Chamoles, Fundación para el Estudio de Enfermedades Neurometabólicas (FESEN), Buenos Aires,
22 Sección de Enfermedades Neuromusculares, Hospital Británico, Buenos Aires,
23 Servicio de Genética Médica, Laboratorio Diagnóstico de Genética y Biología Molecular, Sanatorio Allende, Laboratorio de Biología Celular y Molecular, Fundación Allende, IRNASUS-CONICET, Universidad Católica de Córdoba, Córdoba,
24 Servicio de Neumonología, Hospital Escuela de Corrientes, Corrientes,
25 Consultora del Departamento de Neuropatología, Instituto de Investigaciones Neurológicas-FLENI y Hospital Nacional de Pediatría Prof. Dr. Juan P. Garrahan, Buenos Aires, Argentina
† Fallecido el 4 de febrero de 2017
Dirección postal: Ernesto A. Fulgenzi, Servicio de Neurología, Hospital General de Agudos Dr. Ignacio Pirovano, Av. Monroe 3555, 1428 Buenos Aires, Argentina
e-mail: efulgenzi@intramed.net
Resumen
La enfermedad de Pompe (EP) es un desorden metabólico autosómico recesivo infrecuente, producido por la ausencia o deficiencia de la enzima lisosomal alfa-glucosidasa ácida en los tejidos de los individuos afectados. Se considera enfermedad de Pompe de inicio tardío (EPIT) en aquellos individuos de más de un año de edad al comienzo de los síntomas. El objetivo del presente consenso es el de actualizar las pautas y recomendaciones para un correcto tratamiento de los pacientes con EPIT, tomando como referencia los lineamientos del Consenso Argentino para el diagnóstico, seguimiento y tratamiento de la enfermedad de Pompe publicado en el año 2013. Se organizó un consenso que reunió profesionales con experiencia en la EP en las áreas de clínica médica, diagnóstico de laboratorio, neuropatología, neumonología, nutrición, neurología, enfermedades metabólicas, enfermedades neuromusculares y rehabilitación. Se realizó una actualización de la bibliografía sobre EPIT, con especial atención en las publicaciones relevantes de los últimos cuatro años. Los términos finales del documento fueron consensuados por todo el grupo de trabajo. Cada participante proporcionó su declaración de conflicto de intereses. El resultado es una actualización del último Consenso Argentino para la EP, con particular enfoque en su forma de comienzo tardío. Tratándose de una afección infrecuente, en la que los datos disponibles son limitados, las presentes recomendaciones deben ser consideradas como opinión de expertos.
Palabras clave: Alfa-glucosidasa ácida; Consenso; Deficiencia de maltasa ácida lisosomal; Enfermedad de Pompe; Glucogenosis tipo II; Terapia de reemplazo enzimático.
Abstract
Argentine consensus on late-onset Pompe’s disease
Pompe’s disease (PD) is an infrequent metabolic autosomic recessive disorder produced by the lack or deficiency of the acid alpha-glucosidase lysosomal enzyme in tissues of involved individuals. Delayed-onset PD is considered whenever symptoms onset start after one year of age. We present an update of the recommendations for the management of delayed-onset PD, taking as reference the guidelines from the Argentine Consensus for diagnosis, treatment and follow-up of PD published in 2013. The present consensus gathered several experts in PD in the areas of internal medicine, laboratory diagnosis, neuropathology, pulmonology, nutrition, neurology, metabolic and neuromuscular disorders as well as rehabilitation to perform an update of the literature of delayed-onset PD, with special attention on relevant information published within the last 4 years. The entire working group approved the final version of the consensus. Each participant provided a declaration of conflict of interest. As a result, it is an update of the previous Argentine PD Consensus with focus on the delayed-onset presentation of the disease. Being such infrequent disorder, available data were rather limited and thus, the recommendations represent expert opinions.
Key words: Alfa-glucosidase; Consensus; Acid lisosomal maltase deficiency; Pompe’s disease; Type II glycogenosis; Enzyme replacement therapy.
Las enfermedades de depósito lisosomal son poco frecuentes, en las mismas ocurre una acumulación anormal de sustratos dentro de los lisosomas, lo que produce diferentes trastornos en la estructura y función de los tejidos afectados1. Se estima que se presentan en conjunto en 1/7000-8000 recién nacidos vivos, aunque es probable que estén subdiagnosticadas, ya que existe en general escaso conocimiento sobre las mismas2, 3.
La enfermedad de Pompe (EP), también conocida como glucogenosis tipo II o deficiencia de maltasa ácida (OMIM # 232300), fue la primera enfermedad de depósito lisosomal descripta. Se produce por un trastorno autosómico recesivo que lleva a la ausencia o marcada deficiencia de la enzima lisosomal alfa-glucosidasa ácida (AGA) (EC 3.2.1.20)4.
La AGA lisosomal cataliza la degradación del glucógeno a glucosa y, cuando la actividad de la enzima es deficiente éste se acumula en el lisosoma. Al romperse los lisosomas el glucógeno se vuelca al citoplasma, afectando así a los elementos contráctiles de la célula del músculo esquelético, liso y cardíaco5. Esto conduce a hipotonía y debilidad muscular, por lo que la EP es también una miopatía metabólica dentro de las enfermedades neuromusculares (ENM)6,7.
La tasa de acumulación de glucógeno y, por lo tanto, la gravedad de los síntomas, depende principalmente de la actividad enzimática residual, que suele ser inferior al 1% de la normal en los niños menores de 1 año, hasta el 10% en la forma juvenil y menor al 40% en adultos. El fenotipo de cada paciente está determinado además por factores nutricionales, el tipo de fibra muscular afectada, la actividad física, la edad de inicio de la enfermedad y modificadores genéticos aún no bien comprendidos4,6,8.
La EP es un desorden multisistémico, con un amplio espectro de fenotipos clínicos y grados variables de progresión, síntomas de inicio y niveles de afectación de los distintos órganos9. Se reconocen dos extremos: una forma de inicio infantil y rápida progresión, y otra de inicio tardío y progresión más lenta. Se diferencian así dos categorías muy amplias, la EP infantil y la EP de inicio tardío (EPIT)10. En este consenso se discutirán específicamente aspectos de la EPIT.
Si bien la incidencia global de la EP es variable, la misma se estima en 1: 40 000 según el cálculo de incidencia/prevalencia de mutaciones en la población general6,11. Sin embargo, estudios prospectivos estiman que podría ser tan alta como 1:900012. Teniendo en cuenta la incidencia esperada, la EP es una entidad sub-diagnosticada.
El objetivo del presente consenso es actualizar los conocimientos y las pautas para un correcto diagnóstico, evaluación, manejo y tratamiento de los pacientes con EPIT, tomando como referencia el Consenso Argentino para el diagnóstico, seguimiento y tratamiento de la EP realizado en el año 201313.
Fisiopatología
La enzima lisosomal AGA cataliza la hidrólisis de los enlaces glucosídicos α 1-4 y α 1-6 de la molécula de glucógeno a pH bajo (4.0 a 5.0). Su deficiencia produce acumulación del mismo en múltiples tejidos.
La ruptura de la membrana lisosomal provoca la liberación del glucógeno acumulado, así como la extravasación del contenido del lisosoma al citosol. La acumulación del glucógeno es tanto intra como extra lisosomal14-16. Este depósito lisosomal y secundariamente citoplasmático en el tejido muscular por extravasación desde los lisosomas es un marcador característico de la enfermedad17.
Además de este fenómeno de acumulación existen evidencias de otros mecanismos de daño tisular, entre ellos una desregulación de la autofagia (proceso intracelular por el cual las macromoléculas y organelas son llevadas hacia los lisosomas para su degradación y reciclado)18. Esto provocaría una acumulación progresiva de los autofagosomas, que alteraría el aparato contráctil de las fibras musculares19, 20.
La EP se considera actualmente una enfermedad multisistémica, que incluye también compromiso hepático, aunque en la EPIT el mismo no llega a ser tan grave como para manifestarse clínicamente. Es posible que ante la necesidad de mantener los niveles de glucemia los procesos de degradación del glucógeno en los hepatocitos sean continuos, por lo que es poco el material que queda internalizado en los lisosomas y no llegan a desencadenarse los procesos de daño celular característicos de las fibras musculares21.
Enfermedad de Pompe de inicio tardío
Se define como EPIT cuando la sintomatología aparece luego del año de vida17. Este término puede ser confuso, ya que la EPIT incluye pacientes cuyo cuadro clínico comienza tanto en la niñez como en la juventud y la adultez, aun la avanzada17. Esta categoría constituye por lo tanto un grupo heterogéneo de individuos con afección preponderante del músculo esquelético y poca evidencia de lesión cardíaca6, aunque están descriptas anormalidades electrocardiográficas y arritmias22. En la EPIT, la actividad enzimática se conserva parcialmente en fibroblastos de la piel, hasta en un 40% de los valores normales6 ,23.
El patrón de afectación muscular y su distribución, asociados a la secuencia de aparición de la debilidad en los distintos grupos musculares, le confiere una característica casi única. A pesar de ello su diagnóstico requiere un alto índice de sospecha clínica, para lo que es necesario un cuidadoso interrogatorio y examen neuromuscular. La Tabla 1 menciona algunas ENM con las que la EPIT puede ser confundida. Se han informado demoras en el diagnóstico de hasta 30 años24.
Tabla 1. Enfermedad de Pompe de inicio tardío, diagnósticos diferenciales

La EPIT se caracteriza por debilidad muscular lentamente progresiva con predominio proximal. Las primeras manifestaciones suelen pasar desapercibidas y afectan a los músculos axiales, abdominales y paravertebrales, que se encuentran comprometidos en forma precoz24-26. Este patrón la distingue semiológicamente del grupo de las llamadas “distrofias de cinturas” y de las miopatías inflamatorias.
La fuerza muscular declina con más rapidez en los miembros inferiores que en los superiores, y los trastornos en la deambulación son el motivo de consulta más frecuente en la EPIT en el adulto26.
Los músculos del compartimiento posterior del muslo y los glúteos están característicamente afectados, mientras que los cuádriceps preservan la fuerza por más tiempo9, 25. Los músculos distales se conservan hasta un estadio bastante más avanzado afectándose en menos de un 10% de los casos27.
En los miembros superiores la atrofia de músculos periescapulares puede ser importante en algunos casos, con aparición de escápulas aladas. El bíceps braquial y los músculos distales están en general muy preservados hasta avanzada la enfermedad.
Los flexores del cuello están en general comprometidos tempranamente9. Esto puede asociarse a escoliosis y espina rígida28-30. La debilidad en la lengua está siempre presente, aunque solo en ocasiones provoca disartria31.
La ptosis palpebral uni o bilateral sin compromiso de músculos extraoculares, inicialmente informada en casos aislados, estaría presente en casi un cuarto de los pacientes en nuestro medio32-34.
La disfagia puede aparecer en estadios avanzados23,32. En la primera infancia se puede observar retraso en el desarrollo motor y trastornos en el crecimiento, incluyendo dificultad en la ganancia de peso en los adolescentes.
Una de las marcas o características distintivas de la enfermedad es el compromiso precoz del diafragma, lo que lleva a una falla respiratoria relativamente temprana, aun en pacientes capaces de deambular en forma independiente. La insuficiencia respiratoria es la principal causa de morbilidad y mortalidad en esta enfermedad, y puede ser la manifestación inicial en hasta un tercio de los casos25, 35, 36.
La edad del deceso varía desde la primera infancia hasta la adultez avanzada, en función de la tasa de progresión de la enfermedad, el grado de compromiso de los músculos respiratorios y la presencia de otras comorbilidades6, 37.
Si bien las manifestaciones neuromusculares son las dominantes en la EPIT, puede existir compromiso de otros órganos. Alteraciones en las arterias del sistema nervioso central como dolicoectasia basilar, dilatación de arteria carótida interna y aneurismas de la arteria basilar, cerebral media o carótida interna son hallazgos incidentales frecuentes, y se encuentran raramente luego de hemorragias o microhemorragias parenquimatosas38-40. El hipotiroidismo es un hallazgo común, y se han descripto casos que presentaron hipoacusia, dolor neuropático o disfunción autonómica secundarios a compromiso de fibras nerviosas finas, aunque se desconoce la prevalencia real de este cuadro38, 41, 42.
Además de la signo-sintomatología descripta hasta este punto, síntomas más leves o inespecíficos como mialgias, dolor lumbar, fatigabilidad e intolerancia al ejercicio son comunes en estadios iniciales o preclínicos de la EPIT, y pueden preceder por años a este cuadro sin que el diagnóstico haya sido siquiera sospechado24,25. La variabilidad de la presentación clínica en esta enfermedad es de hecho tan marcada que incluye la existencia de individuos asintomáticos43.
Compromiso respiratorio
Los síntomas respiratorios pueden ser el motivo de consulta inicial en el 30% de los casos con EPIT25, 36, 44. La insuficiencia respiratoria es habitual en el curso de la enfermedad, y puede ser de comienzo agudo o insidioso. El compromiso de los músculos respiratorios constituye la causa más común de muerte prematura en estos enfermos35,45.
Las manifestaciones respiratorias son consecuencia de una combinación en grado variable de la debilidad de los músculos inspiratorios (en particular del diafragma), de los músculos espiratorios, de los músculos de inervación bulbar, de los trastornos respiratorios durante el sueño (TRS)36,44,46, y de las alteraciones del control respiratorio47-49.
El compromiso de los músculos esqueléticos y respiratorios no progresa del mismo modo. Un paciente que deambula puede necesitar ventilación nocturna. Esto es una característica de la enfermedad24,35,50.
Los TRS son diversos y en general pueden ser compartidos con otras ENM con compromiso respiratorio, pero a diferencia de ellas, el compromiso temprano del diafragma puede producir alteraciones nocturnas marcadas con una capacidad vital forzada (CVF) en sedestación relativamente preservada35, 46, 50-57.
Los TRS pueden ser dependientes de la EPIT o no estar relacionados. Dicha diferenciación no siempre es sencilla de establecer. Además de la detección de TRS, interesa en particular establecer la presencia de hipoventilación nocturna. La misma puede ser precoz y desproporcionada al compromiso funcional respiratorio si coexisten parálisis diafragmática y anomalías del control de la ventilación47,58.
Evaluación clínica respiratoria
La debilidad de los músculos respiratorios puede manifestarse como ortopnea, disnea de esfuerzo, de reposo o disnea durante la inmersión en agua hasta el xifoides57. Aproximadamente la mitad de los pacientes adultos refieren disnea de esfuerzo al inicio del padecimiento59.
La presencia de respiración paradojal del abdomen, asociada con ortopnea, es sugestiva de paresia o parálisis diafragmática, un hallazgo que puede ser la expresión inicial de la enfermedad35, 52, 56, 60-63.
Las infecciones respiratorias altas o bajas (traqueobronquitis y neumonía) a repetición son manifestaciones precoces del compromiso del sistema respiratorio, especialmente de la debilidad de los músculos espiratorios, de la tos y de la deglución58.
Todos los pacientes con EPIT deben tener una historia detallada de las características del sueño al momento del diagnóstico y durante el seguimiento51, 64. La presencia de cefaleas matutinas, hipersomnia diurna, fatiga, deterioro intelectual, roncopatía, episodios de apnea o inquietud durante el sueño, obliga a pensar en los TRS25, 52, 53, 56, 57, 65.
Diversos cuestionarios para identificar TRS en las ENM pueden resultar de utilidad. Sin embargo uno solo, el Sleep-Disordered Breathing in Neuromuscular Disease Questionnaire, ha sido validado para pacientes con ENM y parálisis diafragmática66.
El examen físico respiratorio debe ser realizado en sedestación y en decúbito dorsal. Es de fundamental importancia la detección de debilidad de los músculos de inervación bulbar, la presencia de macroglosia, la utilización de músculos accesorios de la respiración, de movimientos respiratorios anormales, y en particular la presencia de respiración paradojal del abdomen o hallazgos constitucionales que predispongan a TRS57,66. Las radiografías de tórax se deben obtener al momento del diagnóstico y cada vez que haya algún cambio o deterioro clínico57.
Evaluación funcional respiratoria
El examen de la función respiratoria debe ser efectuado en forma periódica10, 14, 63, 67. Se recomienda al menos una evaluación respiratoria anual en la EPIT, independientemente del grado de compromiso de la función muscular periférica68. La evaluación funcional respiratoria debe estar dirigida a la valoración del trastorno restrictivo y de la función del diafragma, a la detección de tos débil y la presencia de TRS e hipoventilación alveolar57.
En los pacientes con EPIT la evaluación de la función pulmonar debe incluir la medición de la medición de la CVF, el volumen espiratorio forzado en 1 segundo (VEF1), la simple determinación del Flujo Espiratorio Pico (FEP) y la saturación de O2 por oximetría de pulso. Completan la evaluación la presión inspiratoria máxima (PImax), la presión espiratoria máxima (PEmax), la determinación de la PCO2 al final de la espiración y los gases arteriales25, 54, 69.
La simple determinación de la CVF en decúbito dorsal es ineludible, ya sea para orientación diagnóstica de una ENM en estudio o para control evolutivo de la EPIT ya diagnosticada68. La CVF sentado puede hallarse dentro de valores normales aun en presencia de una CVF en decúbito dorsal reducida68, 70. Se acepta como normal una caída de hasta el 10% de la CVF en decúbito dorsal (habitualmente cae entre 2 y 5 %). Disminuciones mayores al 10% sugieren debilidad diafragmática y una reducción mayor del 30% es altamente sugestiva de debilidad grave o parálisis bilateral68.
La PImax y la PEmax son más sensibles que la CVF para detectar debilidad muscular respiratoria y permiten discriminar la presencia de debilidad inspiratoria y espiratoria por separado18, 57, 68, 69, 71. El FEP es otro parámetro que permite la evaluación objetiva de la fuerza muscular espiratoria en ausencia de obstrucción bronquial58. Este último parámetro se utiliza para definir cuándo se debe comenzar la asistencia de la tos. La estimación conjunta de la saturación arterial de O2 mediante oximetría de pulso y del CO2 arterial por medio de la capnografía en aire espirado puede limitar la utilización de la punción arterial58.
La CVF y la disfunción diafragmática son dos importantes predictores de la insuficiencia respiratoria y de la hipoventilación sostenida durante el sueño52, 60, 70.
En publicaciones recientes, en series de pacientes con EPIT, se observó hipercapnia desproporcionada al grado de debilidad muscular respiratoria y del defecto mecánico. En esos pacientes se halló una respuesta central deprimida a la re-inhalación de CO247,72. Estos trabajos concluyeron que en sujetos con EPIT el impulso central deprimido puede contribuir a la retención de CO2. La repercusión clínica de este fenómeno debe ser evaluada47.
Trastornos respiratorios del sueño
Los cambios fisiológicos que ocurren durante el sueño en sujetos normales se tornan críticos en presencia de debilidad de los músculos respiratorios73. La identificación de anormalidades ventilatorias durante el sueño es importante para iniciar el soporte ventilatorio apropiado35, 46, 57, 74.
Los TRS que se presentan en los enfermos con EPIT incluyen la hipoventilación sostenida y la apnea obstructiva del sueño50, 65. Debido a la desproporcionada debilidad diafragmática se debe considerar el monitoreo de los pacientes con síntomas compatibles con TRS aun cuando la CVF en posición sentado se encuentre dentro de los parámetros normales35, 46, 57. Existe una distribución bimodal de estos trastornos, con predominio de eventos obstructivos en etapas tempranas y eventos pseudocentrales relacionados al sueño REM en estadios más avanzados de la EPIT. Los primeros están asociados a características propias de la enfermedad que facilitan la obstrucción de la vía aérea, como disfunción de los músculos de la lengua y músculos bulbares, mientras que los eventos pseudocentrales resultan de la incapacidad de los músculos respiratorios (fundamentalmente el diafragma) para generar la presión negativa necesaria para colapsar la vía aérea35,56.
La saturometría nocturna es una herramienta diagnóstica sencilla y de gran utilidad. Se han propuesto diversos criterios de desaturación nocturna: 1) Más de 5 minutos consecutivos con una SatO2< 88%. 2) Más del 30% de la noche con SatO2 < 90%75, 76.
La polisomnografía (PSG) es la prueba de laboratorio más importante para la evaluación de los pacientes con hipersomnia y sospecha de TRS. Es una herramienta programada para interpretar las apneas e hipopneas debidas a acontecimientos centrales u obstructivos, más que a debilidad de los músculos respiratorios77. Presenta baja sensibilidad para determinar la presencia o no de hipoventilación nocturna continua. La eficacia de la ventilación nocturna solo puede conocerse mediante la determinación de gases en sangre arterial. La PSG con oximetría de pulso y capnografía transcutánea mejora la sensibilidad para la detección de hipoventilación nocturna y su uso es aconsejado57, 78.
Cabe destacar que puede resultar dificultosa la distinción entre TRS por debilidad diafragmática o por alteraciones en la vía aérea superior. Los estudios con mediciones acopladas de PSG más presión transdiafragmática lo demuestran52, 56, 79, 80. Los estudios de PSG deberían ser interpretados en el contexto clínico de los pacientes, con sus datos funcionales (debilidad de los músculos respiratorios y caída de la CVF en decúbito dorsal) y gasométricos.
El tratamiento de los TRS en pacientes con EPIT y otras ENM requiere de un neumonólogo con experiencia en estas enfermedades, así como de un equipo multidisciplinario compuesto esencialmente por kinesiólogos respiratorios80.
Insuficiencia respiratoria
Los dos aspectos centrales en el cuidado respiratorio son 1) la detección y asistencia de la tos débil y 2) la detección y el tratamiento de la hipoventilación.
La asistencia de la tos débil previene complicaciones respiratorias infecciosas, reduciendo la necesidad de asistencia ventilatoria. Además, constituye uno de los pilares en el tratamiento de la insuficiencia respiratoria aguda junto a la utilización de ventilación no invasiva.
La insuficiencia ventilatoria (hipoventilación) en pacientes con EPIT se desarrolla debido a la debilidad muscular respiratoria progresiva, con o sin TRS, aun en pacientes ambulatorios, y es la causa más común de muerte en estos casos51,59. El rol relativo de los trastornos del control de la ventilación recientemente descriptos debe ser mejor definido47.
El diagnóstico definitivo de insuficiencia respiratoria es gasométrico, es decir mediante la determinación de gases arteriales (los gases venosos no son confiables). Pero para valorar el desarrollo de insuficiencia respiratoria se recomienda estimar de rutina el intercambio de gases realizando una oximetría de pulso y una capnografía. Si son normales, no resultaría necesario evaluar gases en sangre arterial. Sin tratamiento, la insuficiencia respiratoria crónica puede progresar hasta el desarrollo de cor-pulmonale, aunque esto es infrecuente.
Vacunación
Además de las vacunas regulares, todos los pacientes deben recibir las vacunas correspondientes a neumococo e influenza10.
Manejo de las secreciones
Para facilitar la eliminación de las mismas se deben utilizar técnicas manuales y/o mecánicas de tos asistida81-83.
Fármacos
El tratamiento de las infecciones pulmonares debe ser precoz y intenso10, 82.
Asistencia ventilatoria
La presencia de parálisis diafragmática y ortopnea constituyen claras indicaciones de asistencia ventilatoria no invasiva nocturna. No debe ser el oxígeno el tratamiento de la hipoventilación alveolar pura, es decir, sin anomalías del intercambio gaseoso asociadas. El tratamiento debe dirigirse al trastorno respiratorio subyacente que produce hipoxemia.
En los pacientes con EPIT y apneas obstructivas del sueño, habiéndose descartado hipoventilación nocturna y diurna, el tratamiento puede limitarse a la presión positiva continua (CPAP). En pacientes con hipoventilación nocturna, el tratamiento es ventilación no invasiva con presión positiva bi-nivelada (Bi-PAP)50, 52. La modalidad de Bi-PAP debe considerarse si la PCO2 es mayor a 45 mmHg, si la CVF en posición supina es <50% de lo previsto, si la fuerza inspiratoria negativa es <60 cmH2O, o si la SatO2 es <88% (por 5 minutos seguidos) durante el sueño84, 85. Excepto por la primera indicación (PCO2 alta), es necesario evaluar en estudios prospectivos el grado de adherencia a estas recomendaciones.
Si el tratamiento con Bi-PAP no logra corregir la hipoxia y la PaCO2 es normal, se puede utilizar oxígeno suplementario, cuidando su tasa de administración junto con el nivel arterial de dióxido de carbono, para evitar la hipercapnia inducida por oxígeno86. En casos de necesidad de O2 suplementario con normocapnia, se debe considerar la presencia de enfermedad pulmonar concomitante.
Diversos trabajos demostraron que la utilización de terapia de reemplazo enzimático (TRE) puede permitir la estabilización de la función respiratoria y motora, y que este beneficio se mantiene en el tiempo87-92, lo que ha sido confirmado mediante revisión sistemática93. La Tabla 2 resume las principales recomendaciones para el tratamiento de los desórdenes respiratorios en la EPIT.
Tabla 2. Recomendaciones para el manejo y tratamiento de los desórdenes respiratorios en la enfermedad de Pompe de inicio tardío

Métodos de diagnóstico
Ante la sospecha clínica pueden utilizarse diversas pruebas de laboratorio para demostrar la disminución de la actividad enzimática de la AGA y/o la mutación del gen responsable que pueda originarla.
La investigación de actividad enzimática en gota de sangre seca es por su accesibilidad y disponibilidad el primer test diagnóstico al que se debe recurrir. El resultado debe ser confirmado en un segundo ensayo sobre otro tejido: linfocitos, leucocitos, músculo. El diagnóstico sobre fibroblastos puede brindar información sobre el pronóstico de la enfermedad a partir de la actividad enzimática residual encontrada.
Ante un resultado no concluyente o negativo es necesario efectuar una biopsia muscular. Esta podrá aportar elementos que apoyen la hipótesis diagnóstica o será útil para establecer un diagnóstico alternativo14.
Diagnóstico de sujetos en riesgo
Teniendo en cuenta el carácter recesivo de la enfermedad, y la posibilidad de que la misma se mantenga silente por mucho tiempo, es importante que los familiares en riesgo de los pacientes con diagnóstico confirmado, en especial los hermanos, sean estudiados para descartar su afectación y decidir luego de la evaluación clínica la necesidad de TRE.
Análisis de la actividad enzimática
La actividad de AGA medida en cultivo de fibroblastos obtenidos de una biopsia de piel es considerada el gold standard para el diagnóstico de EP en pacientes sintomáticos. Brinda información valiosa en cuanto a la actividad enzimática residual y permite discriminar formas graves y moderadas. Por ser un método lento (el diagnóstico puede demorar hasta 6 semanas) e invasivo, se han desarrollado otros ensayos para medir la actividad de AGA en sangre94.
La actividad de la AGA también se puede medir en linfocitos purificados, músculo, leucocitos o gotas de sangre seca sobre papel de filtro (GSPF), pero en estos dos últimos tejidos se corre el riesgo de que la isoenzima maltasa-glucoamilasa interfiera en la medición de la actividad enzimática realizada a pH ácido5. El uso de acarbosa, que inhibe la isoenzima maltasa-glucosamilasa, ha permitido aumentar la selectividad del método y obtener así resultados confiables17, 23, 95. La detección de la actividad enzimática se puede realizar por métodos fluorométricos o por espectrometría de masa en tándem96. Las muestras son susceptibles a condiciones ambientales extremas durante el envío, conservación y transporte, por lo que si no se toman las debidas precauciones la actividad enzimática se verá alterada, dando la posibilidad de resultados falsos positivos o no concluyentes.
Las GSPF pueden ser obtenidas fácilmente y enviadas desde lugares remotos hasta centros de referencia para su posterior análisis, siendo especialmente útiles para la detección rápida en neonatos y niños94. La recolección de la muestra es un paso importante en la seguridad y eficiencia de la determinación. Un video de la técnica de recolección de GSPF está disponible en línea (Fundación para el Estudio de las Enfermedades Neurometabólicas, 2011). La fecha de la toma siempre debe tenerse en cuenta en la tarjeta para ayudar en la interpretación de los resultados. Las tarjetas deben ser secadas en posición horizontal a temperatura ambiente no superior a 25 °C durante al menos 4 horas y almacenadas a 4 °C después del secado.
El método de espectrometría de masa en tándem, desarrollado para su uso en programas de screening o tamizado neonatal, tiene potencial para ser incorporado a futuro en un panel de estudios de enfermedades lisosomales97.
Biopsia muscular
Si bien en este espacio no consideramos en particular la forma infantil de la EP, es importante tener en cuenta que la misma presenta en la biopsia muscular una grave miopatía vacuolar con acumulación de glucógeno, evidente con técnica de PAS (periodic acid-shiff). Existe disminución de miofibrillas, observable con miosina ATPasa. Las vacuolas son positivas con fosfatasa ácida (marcador lisosomal y de autofagia). Ambos tipos de fibras pueden estar comprometidas. A nivel ultraestructural se suele observar glucógeno intra y extra-lisosomal, distorsión de la estructura con disminución notable de miofibrillas y vacuolas autofágicas (Figs. 1 y 2).

Fig. 1. Biopsia muscular en paciente con enfermedad de Pompe de inicio tardío. Fibras musculares con variaciones de diámetro y contornos redondeados con vacuolas de distinto diámetro, con material PAS (periodic acid-shiff) positivo, fosfatasa ácida positiva. A) Hematoxilina eosina, B) PAS, C) Tricrómico Gomori y D) Fosfatasa ácida
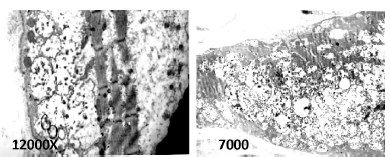
Fig. 2. Biopsia muscular en paciente con enfermedad de Pompe de inicio tardío. A nivel ultraestructural se observa marcada disminución de miofibrillas y material granular correspondiente a glucógeno intralisosomal (en el interior de vacuolas limitadas por membrana) y extralisosomal (libre).
En casos de EPIT, los hallazgos pueden presentar un rango amplio de variación. Las biopsias pueden ser normales, tener cambios mínimos detectables, como una vacuola lineada o no lineada fosfatasa ácida positiva, o bien características graves similares a las descriptas en la forma infantil. Puede observarse aislada fibra necrótica y se ha notificado un patrón neurogénico en algunos casos33, 98-100.
Se ha informado componente inflamatorio en algunos casos de EP. Por lo tanto, la presencia de inflamación en una biopsia no debe excluir esta enfermedad, aun cuando la sospecha clínica sea de miopatía inflamatoria.
Los hallazgos dependen del músculo biopsiado. Es esencial evaluar la biopsia de un músculo comprometido clínicamente. Hay que considerar que la positividad de la biopsia no solo depende del músculo biopsiado sino también del momento evolutivo de la enfermedad98, 99. Las Figuras 1 y 2 muestran los hallazgos anatomopatológicos característicos en esta enfermedad.
En conclusión, la biopsia muscular puede ser característica, pero una biopsia normal, inespecífica o inflamatoria no excluye EP6, 98, 99, y una con hallazgos mínimos, como una sola vacuola lineada o no lineada fosfatasa ácida positiva, debe ser estudiada con microscopía electrónica y controlada la AGA en papel de filtro antes de proseguir con la investigación de otro tipo de miopatías. La biopsia muscular puede ser de importancia decisiva en aquellos casos en donde los valores de las otras determinaciones, como el estudio en gota de sangre seca o los dosajes, arrojen resultados dudosos o ambiguos, en el contexto de un cuadro clínico compatible, tanto para su evaluación morfológica como para el dosaje de AGA sobre el músculo.
Estudios moleculares (ADN)
El gen AGA contiene 20 exones, y solo el primero no es codificante. Se localiza en el cromosoma 17q25101. El ADN codificante (ADNc) codifica un polipéptido de 952 aminoácidos incluyendo un péptido señal de 27 aminoácidos. En el Centro de Pompe (www.pompecenter.nl) hay descriptas más de 500 variantes de secuencia en el gen AGA, de las cuales más de 350 presentan algún grado de patogenicidad, aproximadamente 90 variantes fueron clasificadas como mutaciones no patogénicas y más de 90 variantes fueron clasificadas de significado incierto (última actualización: mayo 2016). Las alteraciones incluyen mutaciones sin sentido, con cambio de sentido, mutaciones que afectan el splicing y deleciones/inserciones, entre otras.
Existen mutaciones muy frecuentes, como la mutación intrónica c.-32-13T›G, que afecta el splicing y da cuenta de casi el 50% de las alteraciones halladas en pacientes caucásicos con EPIT10,101.
Las correlaciones genotipo-fenotipo no se hallan bien definidas, aunque la presencia de mutaciones sin sentido en los dos alelos del gen, es decir mutaciones que producen una proteína trunca, está casi siempre asociada con EP clásica infantil. Otros factores (genéticos y ambientales) pueden influir en el fenotipo, lo que sugiere que pacientes con las mismas mutaciones en el gen AGA pueden presentar fenotipos clínicamente diferentes102.
El análisis de las mutaciones del gen AGA es relevante para el diagnóstico prenatal, siempre que el genotipo del paciente índice sea conocido. También resulta útil para confirmar el diagnóstico en pacientes con EPIT y para la identificación de individuos portadores, cuando una mutación familiar es conocida (si se sabe cuáles son las mutaciones familiares, el examen molecular es el estudio ideal).
La presencia de las mutaciones c.1726G>A (p.G576S) y c.2065G>A (p.E689K) en el mismo alelo (en cis) y ambas en homocigosis, fue descripta como pseudodeficiencia de AGA. Los individuos que poseen estas variantes presentan una actividad descendida de AGA in vitro, en rango similar al observado en pacientes con EPIT, pero una actividad in vivo suficiente como para no desarrollar la enfermedad. El estudio genético es la única herramienta para diferenciar inequívocamente a un paciente con EPIT de un individuo con pseudodeficiencia.
El análisis de las mutaciones del gen AGA es relevante además para el asesoramiento genético. Siendo una enfermedad autosómica recesiva, los padres de un individuo afectado son portadores y presentan un riesgo de recurrencia del 25%. De esto se desprende que se debe ofrecer asesoramiento genético a todos los padres de un niño con EP y a todos los adultos con EPIT, obteniendo un árbol genealógico de tres generaciones como mínimo.
Resonancia magnética por imágenes
En los últimos años la resonancia magnética por imágenes (RMI) ha adquirido un destacado rol en el diagnóstico y caracterización de diversas ENM. A pesar de que brinda hallazgos no específicos, permite reconocer patrones que reducen significativamente las posibilidades diagnósticas, determinar el mejor sitio para la toma de una biopsia muscular y controlar la evolución post-tratamiento103. Este método detecta, además, cambios en el tejido muscular que pueden preceder a la aparición de debilidad en el territorio afectado.
Ante un proceso patológico, la respuesta muscular se limita a pocos patrones que son fácilmente visualizados en la RMI, como el edema o la infiltración adiposa (Tabla 3). El músculo normal muestra una señal intermedia en todas las secuencias de RMI (Fig. 3A). Si un músculo o grupo muscular presenta señal incrementada, estamos siempre frente a una afección muscular. Si dicho incremento se presenta en secuencias fluido sensibles con supresión grasa la miopatía se encuentra en estadio agudo y/o subagudo, mientras que cuando lo vemos en T1 el cuadro es crónico (Tabla 3). La semiología muscular en RMI no solo evalúa la señal muscular sino también la distribución de las lesiones y los cambios morfológicos en los músculos afectados. En la EPIT no es necesario inyectar contraste endovenoso. El mejor plano de visualización o “de corte” es el axial, siendo fundamental obtener un segundo plano de utilidad anatómica.
Tabla 3. Patrones visibles en RMI, secuencias en las que son identificadas y estadio evolutivo que representan


Fig. 3. Resonancia magnética por imágenes en paciente con enfermedad de Pompe de inicio tardío. Lesiones pre-clínicas. Se visualizan en secuencias de supresión grasa. A) Axial SPIR que muestra áreas focales de mayor señal en músculos para-espinales (flechas) difícilmente identificables en secuencia de densidad protónica (B).
En pacientes con EPIT es fundamental identificar lesiones pre-clínicas (Figs. 3B y 4) ya que el hallazgo de las mismas puede, eventualmente, convertirse en un argumento a favor del inicio del tratamiento de reposición enzimática aun en pacientes con manifestaciones clínicas mínimas o asintomáticos104. La RMI en estadios más avanzados permite determinar el grado de infiltración adiposa muscular (Fig. 3C) y evaluar el grado de respuesta en pacientes tratados105.

Fig. 4. Resonancia magnética por imágenes en paciente con enfermedad de Pompe de incio tardío. Cortes sagitales T1 de lengua. A) Sujeto sano. La lengua presenta tejido graso con un patrón de distribución triangular o “en abanico” de base dorsal (línea de puntos). B) Paciente con enfermedad de Pompe de inicio tardío. La lengua muestra habitualmente pérdida o alteración en la distribución normal del tejido graso como signo temprano de afectación muscular (flecha) C) Paciente con cuadro avanzado. Se puede visualizar reemplazo total del tejido muscular por tejido graso (asterisco).
La RMI es una herramienta de gran utilidad en el estudio de las ENM, aunque no reemplaza - bajo ningún punto de vista - al examen clínico; de hecho, es fundamental evaluar las imágenes en el contexto clínico de cada paciente.
Otros exámenes complementarios
Enzimas musculares
La creatinquinasa (CK) puede estar elevada, pero no suele sobrepasar valores de 1500 UI o 2000 UI106. Las transaminasas hepáticas pueden estar elevadas en forma persistente, aun con niveles de CK solo discretamente superiores a los normales. La elevación conjunta de estas enzimas se considera muy sensible para detectar el daño muscular en la EP107.
Electromiograma
En las formas infantiles muestra irritabilidad de membrana y actividad denervatoria, siendo características las descargas repetitivas de alta frecuencia, que se consideran secundarias al compromiso del asta anterior. La contracción voluntaria muestra potenciales de unidad motora polifásicos breves y de baja amplitud108.
La mayoría de los pacientes con EPIT presentan hallazgos compatibles con miopatía en músculos proximales. Puede encontrarse irritabilidad de membrana o franca actividad denervatoria, incluyendo descargas repetitivas complejas o descargas miotónicas, en ausencia de miotonía clínica. En ocasiones el electromiograma en miembros es normal, y los hallazgos descriptos se encuentran restringidos a grupos específicos, en especial músculos paravertebrales108-110. Estudios recientes sugieren que debería realizarse una evaluación de rutina de los músculos paravertebrales lumbares ante la sospecha diagnóstica de EPIT103. La Figura 5 resume los pasos recomendados en el diagnóstico de la EPIT.
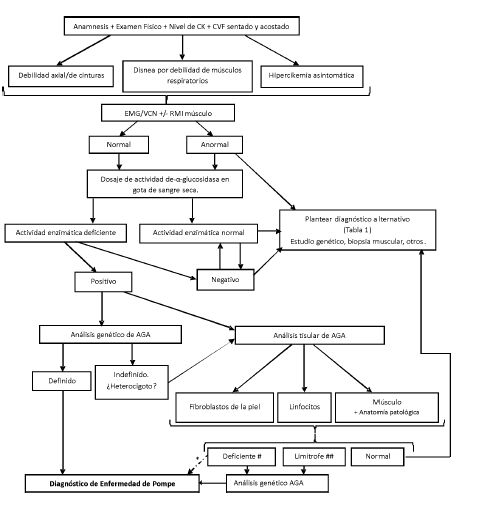
Fig. 5. Algoritmo diagnóstico en la enfermedad de Pompe de inicio tardío. Electromiograma y velocidades de conducción nerviosa (EMG/VCN). # Deficiente: actividad enzimática menor a 30 %. ## Limítrofe: actividad enzimática entre 30-40%
* Consideramos que en un paciente con un cuadro clínico característico de enfermedad de Pompe de inicio tardío asociado a un test positivo en gota de sangre seca y la confirmación del déficit enzimático en otro tejido puede iniciarse el tratamiento de reemplazo enzimático. Sin embargo, es recomendable en todos los casos la confirmación mediante un estudio molecular del gen de AGA.
Tratamiento
Debe entenderse por tratamiento el conjunto de medidas que se toman para curar o atenuar el efecto de la enfermedad. En la EP incluye por un lado el control de los síntomas y las complicaciones derivados de la enfermedad, y por el otro la implementación de una terapéutica específica que procura reponer la enzima faltante o deficitaria (TRE).
El seguimiento de estos pacientes debe involucrar, como en todas las ENM, equipos interdisciplinarios para abordar los distintos problemas clínicos. El equipo de trabajo debe estar integrado por médicos (neurólogo, clínico, neumonólogo, cardiólogo, nutricionista, gastroenterólogo), kinesiólogos, terapeutas físicos, ocupacionales y del lenguaje. Un enfoque multidisciplinario, con la coordinación de un neurólogo, es el más adecuado en estos casos.
Manejo de los trastornos musculoesqueléticos
Los objetivos deben centrarse en la prevención de las complicaciones primarias y secundarias de la enfermedad. De modo similar que para otras ENM se busca lograr la máxima función motora posible. Se debe procurar mejorar la marcha y las transferencias, subir y bajar escaleras, agacharse y levantarse, conservar la función respiratoria y mantener en todo lo posible la independencia para las actividades de la vida diaria. El objetivo final de la rehabilitación es, siempre, mejorar la calidad de vida del paciente.
Como regla general todos los tratamientos físicos deben ser adecuados a cada caso, en relación a su estado actual en particular, y teniendo en cuenta las expectativas futuras10. La inclusión de complementos terapéuticos tales como ejercicios rehabilitatorios de la deglución, foniátricos o terapia ocupacional debe ser individualizada. La rehabilitación debe articularse con otras medidas de soporte como las respiratorias, nutricionales y psicológicas, y el eventual uso de aparatos, ortesis o medios de desplazamiento. Los tratamientos físicos en la EPIT son complementarios y necesarios en forma conjunta con la TRE cuando esta está indicada.
En las ENM en general, el uso de ejercicios contra resistencia ha sido tradicionalmente contraindicado, ante la posibilidad de que ese tipo de práctica produzca un mayor deterioro en los músculos ya comprometidos, acelerando los mecanismos de degeneración111-113. Si bien en la EPIT hay pocos trabajos sobre el efecto de los ejercicios de fortalecimiento, algunos estudios sugieren el uso de ejercicios aeróbicos submáximos114, 115.
Se ha informado que la combinación de ejercicios de resistencia y aeróbicos resulta en la mejoría de la fuerza muscular y en el test de marcha de los 6 minutos en pacientes con EPIT116. Un estudio reciente demostró que esta combinación es beneficiosa, mejorando los valores de fuerza muscular así como también algunas de las pruebas funcionales117. También se ha postulado como favorable el ejercicio aeróbico en otras varias ENM118-120.
La ubicación de los lisosomas entre el aparato contráctil de las fibras musculares podría convertir a esas organelas en más vulnerables al daño producido por las contracciones121. Todos los pacientes con ENM deben ser informados de que el ejercicio intenso implica el riesgo de mayor daño y disminución de la función, y ser instruidos para poder advertir los signos de debilitamiento o fatiga por exceso de ejercicio tales como la aparición de calambres, sensación de pesadez o falta de aire. Hay que extremar los recaudos para evitar la fatiga muscular y que los ejercicios se realicen en condiciones anaerobias. Es importante enseñar al paciente a monitorear su frecuencia cardíaca y respiratoria en relación al ejercicio y brindarle las pautas de alarma necesarias para su manejo. También las estrategias para conservar energía (como programar las actividades del día a día) y obtener ventajas biomecánicas (por ejemplo, el manejo de la velocidad de la marcha, la adecuación del hogar o la elevación de los asientos). El programa de ejercicios debe incluir la caminata, cinta, bicicleta, hidroterapia, y natación. Hay que hacer énfasis en utilizar todos los grupos musculares, agonistas y antagonistas. Con los ejercicios que involucran algún esfuerzo deben tenerse las mismas precauciones que se recomiendan para otras enfermedades neuromusculares de tipo degenerativo122, 123. No existe acuerdo acerca del mecanismo por el cual los ejercicios de resistencia serían beneficiosos en la EPIT. Algunos postulan que el ejercicio muscular continuo disminuiría el acúmulo de glucógeno lisosomal, mejorando así el rendimiento de esos músculos, mientras que otros aseguran que esa mejoría no sería a través de la reducción del sustrato124, 125. La terapia física es más beneficiosa en aquellos pacientes menos comprometidos que en los más afectados126.
Se ha recomendado que los ejercicios empiecen lentamente, permitiendo períodos de descanso, seguidos por un aumento gradual en la intensidad de leve a moderada, hasta llegar a niveles aeróbicos de un 0 a un 70% del esfuerzo máximo, con una frecuencia de 3 a 5 días a la semana10, 36, 112,127.
Es recomendable siempre el control neumonológico durante los programas de rehabilitación ya que, como se mencionó, estos pacientes presentan particular compromiso respiratorio aun en etapas tempranas.
La realización de ejercicios durante la infusión de la TRE no demostró aún adicionar alguna ventaja para los pacientes128.
Las técnicas para tratar y prevenir contracturas y deformaciones articulares son las mismas que las utilizadas para otras ENM e incluyen, por ejemplo, las elongaciones, la utilización de férulas o valvas y el evitar posiciones inapropiadas129.
Debido a la afectación de los músculos espinales lumbares y la tendencia a adoptar posturas compensatorias, se recomienda realizar ejercicios posturales preventivos y enseñar las mejores posturas en la vida diaria con un fin ergonómico. Además de la terapia física y el equipamiento ortésico se debe recurrir a todas las medidas de soporte técnicas, adaptaciones del hogar y elementos para sustentar la movilidad tales como bastones, andadores, soportes para la columna, o el cuello, sillas de ruedas, o scooters. Estas medidas deben ser evaluadas caso por caso por parte del equipo interviniente en el momento adecuado. La Tabla 4 resume los aspectos más importantes del manejo musculoesquelético en la EPIT.
Tabla 4. Recomendaciones para el manejo y tratamiento musculoesquelético de la enfermedad de Pompe de inicio tardío

Escoliosis
La escoliosis constituye una complicación frecuente en muchas ENM130, 131 y ha sido descripta en todas las formas clínicas de la EP30, 132. La debilidad y atrofia progresivas de los músculos del tronco, la utilización prolongada de movimientos y posturas compensatorias sumado a los efectos propios de la gravedad han sido implicados en su desarrollo30,133.
Es más frecuente en pacientes que desarrollan la EP durante la niñez y la adolescencia en comparación con las formas de inicio en la edad adulta, y en pacientes en silla de ruedas30, 133. Se la ha asociado con compromiso de la función respiratoria132. En los casos más graves, la deformidad de la caja torácica puede llevar a la afectación cardiopulmonar, pérdida de la deambulación e imposibilidad de mantener la posición de sentado, además de causar dolor, lo que impacta negativamente en el bienestar y la calidad de vida de los pacientes131.
No hay guías disponibles para el manejo de la escoliosis en pacientes con EP, por lo cual deben seguirse las recomendaciones publicadas para otras ENM con las que se tiene mayor experiencia131, 134.
La presencia de escoliosis debe ser monitoreada y evaluada clínicamente desde las etapas iniciales y confirmada con radiografía. Luego se sugiere continuar con el estudio radiológico periódico cada 6 meses o un año dependiendo de la severidad de la misma30, 51. El tratamiento quirúrgico constituye la única estrategia para prevenir y mejorar la escoliosis grave131, 134.
Osteopenia/osteoporosis
La osteopenia y la osteoporosis han sido descriptas como complicaciones en todas las formas clínicas de la EP y constituyen un factor de riesgo de fracturas tanto en niños como en adultos135-137. Se han informado fracturas en pacientes que aún conservan la deambulación, si bien aquellos con menor movilidad, en silla de ruedas o confinados a la cama presentan una incidencia mayor135, 136.
Una reducción en la fuerza/carga mecánica sobre el sistema óseo y una menor movilidad relacionada con la debilidad muscular han sido propuestas como principales responsables de la menor densidad mineral ósea en pacientes con EP y otras ENM, aunque otros factores podrían resultar influyentes y deben considerarse138-141.
Se recomienda la evaluación con absorciometría de rayos X de energía dual (DEXA) en estos casos, aunque no existe suficiente evidencia para establecer una frecuencia, por lo que el criterio debe ajustarse de manera individual10. En caso de osteopenia u osteoporosis los enfermos deben ser evaluados de forma rutinaria por un especialista.
El uso de vitamina D y calcio se recomienda en aquellos pacientes con un z-score anormal en el DEXA, así como el uso de bifosfonatos de acuerdo a las guías utilizadas para la población general51. La TRE no parece modificar significativamente la densidad mineral ósea, aunque esto no ha sido analizado en grandes cohortes142, 143. Promover la bipedestación en las sesiones de rehabilitación puede ser de utilidad para mejorar la salud mineral ósea. Es también importante la evaluación del riesgo de caídas y el eventual uso de dispositivos de asistencia de marcha para disminuir dicho riesgo51.
Manejo nutricional, alimentario y gastrointestinal
La debilidad de los músculos bulbares es frecuente en pacientes adultos con EPIT. El compromiso de la lengua es habitual, y puede ser evaluado clínicamente31, por métodos cuantitativos144 y por imágenes145 (Fig. 4). En casos con debilidad lingual significativa puede estar afectado el manejo del bolo alimenticio en la fase masticatoria. La disfagia orofaríngea ha sido descripta en pacientes con EPIT146 y su frecuencia y gravedad parecen correlacionarse con el grado de disartria144. Las alteraciones citadas aumentan el riesgo de trastornos secundarios como microaspiraciones y neumopatías.
Síntomas gastrointestinales difusos como diarrea crónica, constipación, molestias o calambres abdominales, plenitud postprandial o saciedad temprana, y pobre ganancia de peso en la adolescencia también han sido descriptos en la EPIT147.
Muy ocasionalmente, en casos con enfermedad avanzada, puede ser necesario proteger al paciente mediante recursos como la sonda nasogástrica o una gastrostomía.
La disfunción digestiva ha sido informada en la literatura, así como su mejoría con la TRE148. El soporte nutricional y alimentario debe estar a cargo de profesionales de la salud idóneos en el tema.
Los pacientes con EPIT requieren evaluación nutricional integral desde el momento de realizado el diagnóstico y controles periódicos a través de toda la evolución de la enfermedad.
La estrategia nutricional requiere conocer la tolerancia y las dificultades en la masticación y la deglución51, y monitoreo de los hábitos alimentarios del enfermo (calidad, cantidad, tipo de alimentos ingeridos, intolerancias, dificultades) y de la familia, para establecer un programa personalizado, efectivo y sustentable de educación y rehabilitación nutricional.
El paciente y su familia deben ser capacitados y/o informados acerca de la elección, combinación y preparación de los alimentos, teniendo en cuenta por ejemplo la frecuencia de ingestas y el tamaño de las porciones, acciones que favorecen la adherencia al tratamiento.
Algunos autores sugieren que podría ser beneficiosa una disminución del contenido graso y de carbohidratos, fundamentalmente los refinados, y el aumento de aminoácidos como fuente energética10, 115, 149-151. Deben considerarse el incremento de la ingesta de la fibra alimentaria soluble e insoluble, y el uso de suplementos dietarios adecuados.
El estado nutricional basal es pronóstico de los resultados de la TRE, el BMI indicado es mayor o igual a 18 kg/m2 y debe ser tomado en cuenta para la indicación de dicho tratamiento152, 153.
Terapia de reemplazo enzimático
El tratamiento con TRE para la EPIT fue aprobado en el año 2006 por la Agencia Europea de Medicamentos y en el 2010 por la FDA y en la actualidad es el único tratamiento farmacológico específico para la EP. La misma abastece de forma exógena la enzima faltante a través de infusiones intravenosas repetidas. La AGA humana recombinante (AGAhr) (Myozyme; Genzyme Corporation) ha demostrado ser eficaz en el tratamiento de los pacientes con EP de inicio precoz y tardío10, 87, 154-158. La dosis recomendada es de 20 mg/kg de peso corporal administrada una vez cada dos semanas por vía endovenosa. Se probaron dosis mayores, de 40 mg/kg de peso cada dos semanas, pero no demostraron un beneficio claro por sobre la dosis habitual, y parecen asociarse con mayor frecuencia a eventos adversos87, 156.
En la EPIT el objetivo principal de la TRE es la prevención de la pérdida adicional de la función muscular87. Los datos indican que la AGAhr tiene un efecto positivo sobre la historia natural de la enfermedad en adultos, y sobre los procesos que llevan a la insuficiencia respiratoria y el deterioro de la deambulación87, 92, 159. En el conjunto de los distintos estudios realizados hasta el año 2012 al menos dos tercios de los pacientes se estabilizaron o mejoraron sus niveles de CK y la función respiratoria y/o muscular luego de la TRE93. El beneficio en la deambulación y la estabilización en la función ventilatoria parecen prolongarse al menos hasta la semana 104 de infusión continuada88. Un estudio reciente de una población de 283 pacientes con EPIT ha demostrado el impacto positivo de la TRE en la sobrevida de estos pacientes91. Los resultados de algunos estudios sugieren un mayor beneficio cuando la TRE se instaura en fase precoz y en pacientes cuya situación clínica basal está mejor conservada160, 161, pero también se ha observado mejoría o estabilización de la enfermedad en pacientes con estadios avanzados o muy discapacitados por la misma162.
La calidad de vida también se ve afectada positivamente con el tratamiento de TRE, como lo indican estudios que miden ese parámetro así como la participación en la vida cotidiana163.
Recomendaciones para el inicio de la terapia de reemplazo enzimático
Teniendo en cuenta el análisis de distintos informes de la literatura y la evidencia disponible sobre la indicación de TRE en pacientes con EPIT51 se pueden distinguir los siguientes grupos (Tabla 5):
Tabla 5. Recomendaciones para el inicio de la terapia de reemplazo enzimático de la enfermedad de Pompe de inicio tardío basadas en el estado y gravedad

Individuos asintomáticos sin signos clínicos objetivos con diagnóstico confirmado por estudios de laboratorio
Ya sea como resultado de estudios de screening familiar o por la mayor disponibilidad de estudios enzimáticos o moleculares, el diagnóstico de individuos pre sintomáticos y asignológicos es creciente164-167. Existe controversia respecto de la iniciación de TRE en este grupo faltando evidencia que confirme que retrasa la aparición de la enfermedad168. Los sujetos en esta condición deben ser examinados cada 6 meses con evaluación de la fuerza muscular (en lo posible cuantitativa) y función respiratoria. La TRE debería iniciarse ante la aparición de síntomas asociados a debilidad muscular o reducción de la CVF que pueda ser atribuida a la enfermedad, es decir, solo cuando se encuentren los primeros signos objetivos de EP. Es importante tener en cuenta que si se detecta la presencia de EP en un determinado sujeto se recomienda encarar la investigación diagnóstica en los hermanos164.
Los estudios por imágenes pueden ser decisivos a la hora de considerar el inicio de la TRE en este grupo de pacientes. Así, en pacientes asintomáticos sin signos clínicos objetivos y con espirometría normal debe considerarse realizar una RMI de músculos, con énfasis en paravertebrales y músculos axiales, para determinar la existencia de compromiso pre-sintomático. En esta situación el comienzo de la TRE debe considerarse caso por caso169.
Pacientes asintomáticos con signos objetivos
Este grupo incluye pacientes con debilidad muscular o reducción de la CVF que puedan ser atribuidas a la enfermedad, y deberían iniciar tratamiento con AGAhr.
Pacientes sintomáticos
La TRE con AGAhr se recomienda para los pacientes sintomáticos que tengan debilidad muscular demostrable en el examen físico o reducción de los parámetros pulmonares en las pruebas de función respiratoria. En caso contrario puede considerarse la TRE si el paciente tiene dificultades en la realización de las actividades de la vida diaria que puedan ser atribuidas a la enfermedad.
Duración de la terapia de reemplazo enzimático
Una vez que se inició la TRE este tratamiento es a largo plazo, tal vez durante toda la vida. Vale la pena destacar que la respuesta individual a la TRE puede variar debido a distintos factores, como el desarrollo de anticuerpos específicos contra la AGAhr, la edad de presentación, la tasa de progresión de la enfermedad, el tipo de fibra muscular, presencia de defectos en la autofagia y el genotipo subyacente8, 10. La discontinuación de la TRE debe considerarse si existen reacciones adversas serias que pongan en peligro la vida del paciente o complicaciones graves derivadas de la instauración de la misma.
Efectos de la terapia de reemplazo enzimático en el músculo esquelético
La mejor respuesta de la TRE sobre el músculo esquelético se ha observado en enfermos tratados de forma temprana, antes de un daño muscular irreversible. No obstante, esta respuesta ha sido más errática que la del músculo cardíaco. Esto podría deberse a que, si bien la captación enzimática en ambos tipos musculares está mediada por receptores de manosa-6-fosfato, el músculo esquelético tendría muchos menos receptores que el corazón, lo que podría redundar en una menor captación de la enzima170, 171. Existen otras teorías que tienden a explicar la menor efectividad de la TRE sobre el músculo esquelético170, 171. A esto se debe añadir que dicho músculo constituye aproximadamente el 40% de la masa corporal total, por lo que el volumen a tratar es mucho mayor que el del músculo cardíaco.
Reacciones adversas de la terapia de reemplazo enzimático
La TRE es en general bien tolerada, y la mayoría de los eventos adversos son leves o moderados93. Aquellos pacientes que presentan una enzima nativa con diferencias significativas con la enzima recombinante administrada, pueden producir anticuerpos contra la enzima exógena, lo que reduce su eficacia y puede ocasionar reacciones adversas ante la infusión. Muchos pacientes también desarrollan tolerancia con el tiempo. En la EPIT se ha visto disminución de anticuerpos anti-AGA en pacientes bajo tratamiento con Myozyme por períodos prolongados172.
A pesar de que la mayoría de las reacciones a la infusión son en general fáciles de tratar, en algunos casos puntuales se han observado reacciones anafilácticas peligrosas, alergia, fiebre y respuestas autoinmunes87, 159. Estas complicaciones pueden aparecer durante el tratamiento o en horas posteriores al mismo154, 156. Las reacciones anafilácticas son una complicación potencialmente grave del tratamiento con cualquier proteína humana recombinante, y se observan en alrededor de un 5% de los casos de EPIT. Los pacientes tratados con AGAhr que desarrollen títulos altos de anticuerpos deben ser monitoreados en forma estrecha45, con un equipo médico siempre disponible durante la infusión.
Suspensión de la terapia de reemplazo enzimático
Se considerará la suspensión del tratamiento luego de evidenciarse en forma objetiva y persistente un deterioro en las evaluaciones mencionadas en la Tabla 6 luego de 52 semanas de tratamiento. Además de estas evaluaciones hay que descartar otras etiologías que puedan justificar el deterioro (hipotiroidismo, déficit de vitamina D), evaluar el título de anticuerpos anti-AGA, la RMI de músculo y eventualmente la biopsia muscular.
Si se decide suspender la TRE deberán continuarse las evaluaciones periódicas. Si luego de suspender la TRE se observa un aumento en la tasa de deterioro se debería considerar el reinicio de la misma. Medidas tales como el aumento de la dosis pueden ser ofrecidas por el médico tratante en función de la evolución del paciente169.
Embarazo y terapia enzimática de reemplazo
No se han reportado efectos adversos o malformaciones congénitas en bebés de madres que han recibido TRE durante el embarazo. Por el contrario, la suspensión de la TRE al inicio del embarazo ha empeorado los síntomas en la madre y produjo aumento de reacciones alérgicas al reiniciar el tratamiento173.
Con respecto al seguimiento en este período, se recomiendan controles por equipo multidisciplinario y se deben realizar evaluaciones respiratorias periódicas. Eventualmente se podría requerir el uso de ventilación no invasiva que luego del parto y el puerperio podría discontinuarse. Se han informado partos por vía vaginal sin complicaciones174.
Evaluación y seguimiento clínico
Escalas de fuerza muscular
Para la evaluación de la fuerza muscular segmentaria se recomienda, en pacientes mayores de 8 años de edad, la escala del Medical Research Council (MRC)175, que consiste en puntuar del 0 al 5 la fuerza ejercida por diferentes grupos musculares.
Escalas de función motora
La evaluación motora más difundida y utilizada en la EP es la prueba de marcha de los seis minutos176, que mide la distancia recorrida en este período. A pesar de ser discutible esta prueba es recomendada internacionalmente. Una importante limitación es que solo es aplicable en pacientes que mantienen la deambulación. También pueden incluirse pruebas cronometradas de la función motora, como las de marchar/correr una distancia de 10 metros, subir y bajar cuatro escalones, y el tiempo requerido para incorporarse desde el piso. Estas pruebas también se han validado en la enfermedad de Duchenne, y pueden utilizarse junto a la prueba de marcha de los seis minutos177. También se recomienda incluir a la escala de fatiga en el seguimiento (edición en español)178.
Escalas de calidad de vida
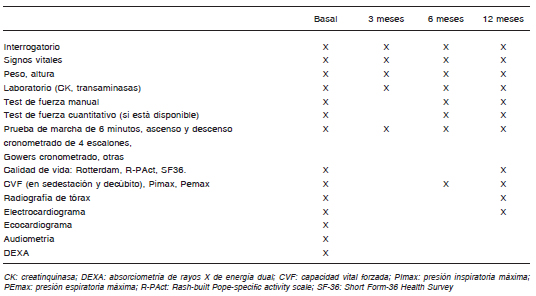
Se ha desarrollado la escala de discapacidad de Rotterdam179 y la escala de actividad específica de la EP construida según el análisis de Rasch (R-PAct). Estas pruebas han sido diseñadas en cohortes de pacientes mayores de 16 años con EPIT. También puede usarse el Short Form 36 (SF 36)181 ampliamente utilizado para diferentes enfermedades, incluida la EP181. La Tabla 6 resume los principales estudios y pruebas para el seguimiento del paciente con EPIT en TRE.
Tabla 6. Protocolo de evaluaciones sugeridas en pacientes con EPIT previa y durante la terapia de reemplazo enzimático
Registro Argentino de la Enfermedad de Pompe
Este registro es un programa multicéntrico, internacional, longitudinal y observacional para pacientes con EP, diseñado para monitorear la evolución natural y resultados de los mismos. Actualmente se encuentra en desarrollo para su aplicación nacional.
Los principales objetivos del registro son: mejorar la comprensión de la variabilidad, progresión y evolución natural de las manifestaciones de la EP; ayudar a la comunidad de médicos especialistas en el desarrollo de recomendaciones para el monitoreo e informe sobre resultados de pacientes, con el objeto de optimizar su atención; caracterizar y describir la población de pacientes con EP como un todo, y evaluar la seguridad a largo plazo y efectividad de las opciones de tratamiento disponibles y medidas de apoyo, incluyendo la TRE con Myozyme® o Lumizyme® (alglucosidasa alfa).
Si bien el protocolo proporciona un Programa de Evaluación Recomendado, que fue desarrollado basándose en el aporte de los médicos de la comunidad internacional con experiencia en el cuidado de pacientes con EP y de entidades reguladoras, cada médico es el único responsable de determinar el cuidado clínico apropiado en cada caso.
Conflicto de intereses: Los Dres. De Vito, Dubrovsky, Barroso, Binaghi, Ferradás, Fulgenzi, Lubieniecki, Politei, Reisin, Rugiero y Taratuto y el licenciado Corderi declaran haber recibido honorarios por clases y/o conferencias por parte del laboratorio Genzyme/Sanofi. Los Dres. Fulgenzi, Lubieniecki, Rugiero y Taratuto declaran haber recibido viáticos para asistencia a Congresos y/o eventos científicos de parte del laboratorio Genzyme/Sanofi. Los Dres. Dubrovsky, Lubieniecki y Rugiero declaran haber recibido subsidios de investigación de parte del laboratorio Genzyme/Sanofi. El Dr. Dubrovsky es miembro del Global Advisory Board for Pompe Disease del laboratorio Genzyme. Los Dres. Berardo, Bettini, Calabrese, Carlés, Chávez, Conti, Di Gennaro, Jáuregui, Medina, Pirra, Rosa, Salutto, Schenone y Sussini declaran no presentar conflicto de intereses.
1. Wilcox WR. Lysosomal storage disorders: the need for better pediatric recognition and comprehensive care. J Pediatr 2004; 144: S3-14. [ Links ]
2. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders JAMA 1999; 281: 249-54. [ Links ]
3. Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet 1999; 105: 151-6. [ Links ]
4. Hers HG. Alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease). Biochem J 1963; 86: 11-6.
5. Thurberg BL, Lynch Maloney C, Vaccaro C, et al. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Invest 2006; 86: 1208-20. [ Links ]
6. Hirschhorn R, Reuser A. The Glycogen storage disease type II: Acid alpha-glucosidase (acid maltase) deficiency. En: Scriver RC, Beaudet AL, Sly WS, Valle D, (eds). Metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001, p 3389–420.
7. Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr 2004; 144: S35-43. [ Links ]
8. Fukuda T, Ahearn M, Roberts A, et al. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol Ther 2006; 14: 831-9. [ Links ]
9. Dubrovsky A, Corderi J, Karasarides T, Taratuto AL. Pompe disease, the must-not-miss diagnosis: A report of 3 patients. Muscle Nerve 2013; 47: 594-600. [ Links ]
10. Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guidelines. Genet Med 2006; 8: 267-88. [ Links ]
11. Martiniuk F, Chen A, Mack A, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet 1998; 79: 69-72. [ Links ]
12. Mechtler TP, Stary S, Metz TF, et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet 2012; 379: 335-41. [ Links ]
13. Dubrovsky A, Fulgenzi E, Amartino H, et al. Consenso Argentino para el diagnóstico, seguimiento y tratamiento de la Enfermedad de Pompe. Neurolarg 2014; 6: 96-113. [ Links ]
14. American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve 2009; 40: 149-60. [ Links ]
15. Raben N, Roberts A, Plotz PH. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol 2007; 26: 45-8. [ Links ]
16. Kuech EM, Brogden G, Naim HY. Alterations in membrane trafficking and pathophysiological implications in lysosomal storage disorders. Biochimie 2016; 130: 152-62. [ Links ]
17. Kishnani PS, Beckemeyer AA, Mendelsohn NJ. The new era of Pompe disease: advances in the detection, understanding of the phenotypic spectrum, pathophysiology, and management. Am J Med Genet C Semin Med Genetics 2012; 160C: 1-7. [ Links ]
18. Raben N, Wong A, Ralston E, Myerowitz R. Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am J Med Genet C Semin Med genetics 2012; 160C: 13-21. [ Links ]
19. Raben N, Takikita S, Pittis MG, Bembi B, Marie SK, Roberts A, et al. Deconstructing Pompe disease by analyzing single muscle fibers: to see a world in a grain of sand. Autophagy 2007; 3: 546-52. [ Links ]
20. Dasouki M, Jawdat O, Almadhoun O, et al. Pompe disease: literature review and case series. Neurol Clin 2014; 32: 751-76. [ Links ]
21. Koeberl DD, Kishnani PS, Chen YT. Glycogen storage disease types I and II: treatment updates. J Inherit Metab Dis 2007; 30: 159-64. [ Links ]
22. Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, Reuser AJ. The genotype-phenotype correlation in Pompe disease. Am J Med Genet C Semin Med genetics 2012; 160C: 59-68. [ Links ]
23. Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 2006; 148: 671-6. [ Links ]
24. Hagemans ML, Winkel LP, Van Doorn PA, et al. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 2005; 128: 671-7.
25. Desnuelle C, Salviati L. Challenges in diagnosis and treatment of late-onset Pompe disease. Curr Opin Neurol 2011; 24: 443-8. [ Links ]
26. Wokke JH, Escolar DM, Pestronk A, et al. Clinical features of late-onset Pompe disease: a prospective cohort study. Muscle Nerve 2008; 38: 1236-45. [ Links ]
27. van der Beek NA, de Vries JM, Hagemans ML, et al. Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study. Orphanet J Rare Dis 2012; 7: 88. [ Links ]
28. Laforet P, Doppler V, Caillaud C, et al. Rigid spine syndrome revealing late-onset Pompe disease. Neuromuscul Disord 2010; 20: 128-30. [ Links ]
29. Kostera-Pruszczyk A, Opuchlik A, Lugowska A, et al. Juvenile onset acid maltase deficiency presenting as a rigid spine syndrome. Neuromuscul Disord 2006; 16: 282-5. [ Links ]
30. Roberts M, Kishnani PS, van der Ploeg AT, et al. The prevalence and impact of scoliosis in Pompe disease: lessons learned from the Pompe Registry. Mol Genet Metab 2011; 104: 574-82.
31. Dubrovsky A, Corderi J, Lin M, Kishnani PS, Jones HN. Expanding the phenotype of late-onset Pompe disease: tongue weakness: a new clinical observation. Muscle Nerve 2011; 44: 897-901. [ Links ]
32. Barnes D, Hughes RA, Spencer GT. Adult-onset acid maltase deficiency with prominent bulbar involvement and ptosis. JR Soc Med 1993; 86: 50. [ Links ]
33. Amato AA. Acid maltase deficiency and related myopathies. Neurol Clin 2000;18: 151-65. [ Links ]
34. Mazia C, Rugiero MF, Dubrovsky A, et al. Ptosis in a cohort of patient with Late Onset Pompe Disease (LOPD) from Argentina. Neurology 2015; 84. [ Links ]
35. Fuller DD, ElMallah MK, Smith BK, et al. The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol 2013; 189: 241-9. [ Links ]
36. Di Rocco M, Buzzi D, Taro M. Glycogen storage disease type II: clinical overview. Acta Myol 2007; 26: 42-4. [ Links ]
37. Hagemans ML, Janssens AC, Winkel LP, et al. Late-onset Pompe disease primarily affects quality of life in physical health domains. Neurology 2004; 63: 1688-92. [ Links ]
38. Filosto M, Todeschini A, Cotelli MS, et al. Non-muscle involvement in late-onset glycogenosis II. Acta Myol 2013; 32: 91-4. [ Links ]
39. Sacconi S, Bocquet JD, Chanalet S, Tanant V, Salviati L, Desnuelle C. Abnormalities of cerebral arteries are frequent in patients with late-onset Pompe disease. J Neurol 2010; 257:1730-3. [ Links ]
40. Sandhu D, Rizvi A, Kim J, Reshi R. Diffuse cerebral microhemorrhages in a patient with adult-onset Pompe’s disease: a case report. J Vasc Inter Neurol 2014; 7: 82-5.
41. Schneider J, Burmeister LA, Rudser K, Whitley CB, Jarnes Utz J. Hypothyroidism in late-onset Pompe disease. Mol Genet Metab Rep 2016; 8: 24-7. [ Links ]
42. Hobson-Webb LD, Austin SL, Jain S, Case LE, Greene K, Kishnani PS. Small-fiber neuropathy in pompe disease: first reported cases and prospective screening of a clinic cohort. Am Jour Case Rep 2015; 16: 196-201. [ Links ]
43. Bembi B, Cerini E, Danesino C, et al. Diagnosis of glycogenosis type II. Neurology 2008; 71: S4-11. [ Links ]
44. Laforet P, Nicolino M, Eymard PB, et al. Juvenile and adult-onset acid maltase deficiency in France: genotype-phenotype correlation. Neurology 2000; 55: 1122-8. [ Links ]
45. Braun NMT, Arora NS, Rochester DF. Respiratory muscle and pulmonary function in polymyositis and other proximal myopathies. Thorax 1983; 38:616-23. [ Links ]
46. Fiorentino G, Annunziata A, Politano L. Sleep breathing disorders and nocturnal respiratory pattern in patients with glycogenosis type II. Acta Myol 2014; 33: 100-3. [ Links ]
47. De Vito EL, Monteiro SG, Aruj PK. Blunted hypercapnic respiratory drive response in subjects with late-onset Pompe disease. Respir Care 2016; 61: 930-5. [ Links ]
48. Anderson TM, Garcia AJ 3rd, Baertsch NA, et al. A novel excitatory network for the control of breathing. Nature 2016; 536: 76-80. [ Links ]
49. Rialp G, Raurich JM, Llompart-Pou JA, Ayestaran I, Ibañez J. Central respiratory drive in patients with neuromuscular diseases. Respir Care 2013; 58: 450-7. [ Links ]
50. Pellegrini N, Laforet P, Orlikowski D, et al. Respiratory insufficiency and limb muscle weakness in adults with Pompe’s disease. Eur Respir J 2005; 26: 1024-31.
51. Cupler EJ, Berger KI, Leshner RT, et al. Consensus treatment recommendations for late-onset Pompe disease. Muscle Nerve 2012; 45: 319-33. [ Links ]
52. Moufarrej NA, Bertorini TE. Respiratory insufficiency in adult-type acid maltase deficiency. South Med J 1993; 86: 560-7. [ Links ]
53. Ward NS, Hill NS. Pulmonary function testing in neuromuscular disease. Clin Chest Med 2001; 22: 769-81. [ Links ]
54. Margolis ML, Howlett P, Goldberg R, Eftychiadis A, Levine S. Obstructive sleep apnea syndrome in acid maltase deficiency. Chest 1994; 105: 947-9. [ Links ]
55. Mellies U, Dohna-Schwake C, Voit T. Respiratory function assessment and intervention in neuromuscular disorders. Curr Opin Neurol 2005; 18: 543-7. [ Links ]
56. Aboussouan LS. Sleep-disordered breathing in neuromuscular disease. Am JRespir Crit Care Med 2015; 191: 979-89. [ Links ]
57. Boentert M, Prigent H, Vardi K, et al. Practical recommendations for diagnosis and management of respiratory muscle weakness in late-onset Pompe disease. Int J Mol Sci 2016; 17:1735. [ Links ]
58. De Vito EL. Sueño en las enfermedades neuromusculares. En: Rabec CA, Franceschini CM, (eds). Trastornos respiratorios del sueño y ventilación mecáncia no invasiva. Buenos Aires, Argentina: Ediciones Journal, 2016, p 135-146. [ Links ]
59. Mellies U, Ragette R, Schwake C, Baethmann M, Voit T, Teschler H. Sleep-disordered breathing and respiratory failure in acid maltase deficiency. Neurology 2001; 57: 1290-5. [ Links ]
60. Raben N, Plotz P, Byrne BJ. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med 2002; 2: 145-66. [ Links ]
61. Kim DG, Jung K, Lee MK, et al. A case of juvenile form Pompe’s disease manifested as chronic alveolar hypoventilation. J Korean Med Sci 1993; 8: 221-4.
62. Keunen RW, Lambregts PC, Op de Coul AA, Joosten EM. Respiratory failure as initial symptom of acid maltase deficiency. J Neurol Neurosurg Psychiatry 1984; 47: 549-52. [ Links ]
63. Domínguez Flores ME, Luna Padrón E, Peñalosa Ochoa L, et al. Guía para el diagnóstico y tratamiento de las alteraciones respiratorias en las enfermedades neuromusculares. Neumol Cir Torax 2011; 70: 1-70. [ Links ]
64. Muller-Felber W, Horvath R, Gempel K, et al. Late onset Pompe disease: clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients. Neuromuscul Disord 2007; 17: 698-706. [ Links ]
65. Perrin C, D’Ambrosio C, White A, Hill NS. Sleep in restrictive and neuromuscular respiratory disorders. Semin Respir Crit Care Med 2005; 26: 117-30.
66. Steier J, Jolley CJ, Seymour J, et al. Screening for sleep-disordered breathing in neuromuscular disease using a questionnaire for symptoms associated with diaphragm paralysis. Eur Respir J 2011; 37: 400-5. [ Links ]
67. van der Ploeg AT. Monitoring of pulmonary function in Pompe disease: a muscle disease with new therapeutic perspectives. Eur Respir J 2005; 26: 984-5. [ Links ]
68. ATS/ERS Statement on respiratory muscle testing. American Thoracic Society/European Respiratory Society. Am J Respir Crit Care Med 2002; 166: 518-624. [ Links ]
69. De Vito EL, Grassino A. Respiratory muscle fatigue: Rationale for diagnostic test. En: Roussos C, editor, The Thorax 2nd ed. New York: Marcel y Dekker Inc, 1995; p 1857-79. [ Links ]
70. Kravitz A, Mackey J, DeArmey S, Kishnani P. Pulmonary function findings in patients with infantile Pompe disease. Proc Am Thorac Soc 2005; 2: A186. [ Links ]
71. Berger J, Skrinar A, Norman BE. Ventilatory dysfunction in late onset Pompe disease. Proc Am Thorac Soc 2005; 2: A788. [ Links ]
72. Berger KI, Chan Y, Rom WN, Oppenheimer BW, Goldring RM. Progression from respiratory dysfunction to failure in late-onset Pompe disease. Neuromuscul Disord 2016; 26: 481-9. [ Links ]
73. Sowho M, Amatoury J, Kirkness JP, Patil SP. Sleep and respiratory physiology in adults. Clin Chest Med 2014; 35: 469-81. [ Links ]
74. Aboussouan LS, Mireles-Cabodevila E. Sleep-Disordered Breathing in Neuromuscular Disease: Diagnostic and Therapeutic Challenges. Chest 2017; 152:880-92. [ Links ]
75. Clinical indications for noninvasive positive pressure ventilation in chronic respiratory failure due to restrictive lung disease, COPD, and nocturnal hypoventilation. A consensus conference report. Chest 1999; 116: 521-34. [ Links ]
76. Levi-Valensi P, Aubry P, Rida Z. Nocturnal hypoxemia and long-term oxygen therapy in COPD patients with daytime PaO2 60-70 mmHg. Lung 1990; 168S: 770-5. [ Links ]
77. Nicolle MW. Sleep and neuromuscular disease. Semin Neurol 2009; 29: 429-37. [ Links ]
78. Weinberg J, Klefbeck B, Borg J, Svanborg E. Polysomnography in chronic neuromuscular disease. Respiration 2003; 70: 349-54. [ Links ]
79. Bach JR, Mahajan K. What is “sleep-disordered breathing” for patients with neuromuscular weakness? Phys Med Rehabil 2007; 88: 1744-5.
80. Finder JD, Birnkrant D, Carl J, et al. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med 2004; 170: 456-65. [ Links ]
81. LCD, for Respiratory Assist Devices (L27228), National Government Services, website. En: https://apps.ngsmedicare.com/applications/Content.aspx?DOCID=21448&CatID=3&RegID=51&ContentID=34363; consultado febrero 2018. [ Links ]
82. Chatwin M, Ross E, Hart N, Nickol AH, Polkey MI, Simonds AK. Cough augmentation with mechanical insufflation/exsufflation in patients with neuromuscular weakness. Eur Respir J 2003; 21: 502-8. [ Links ]
83. Kushida CA, Littner MR, Morgenthaler T, et al. Practice parameters for the indications for polysomnography and related procedures: an update for 2005. Sleep 2005; 28: 499-521. [ Links ]
84. Clinical indications for noninvasive positive pressure ventilation in chronic respiratory failure due to restrictive lung disease, COPD and nocturnal hypoventilation —a consensus conference report. Chest 1999; 116:521-34.
85. Aubier M, Murciano D, Milic-Emili J, et al. Effects of the administration of O2 on ventilation and blood gases in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis 1980; 122: 747-54. [ Links ]
86. van der Ploeg AT, Barohn R, Carlson L, et al. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol Genet Metab 2012; 107: 456-61. [ Links ]
87. van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 2010; 362: 1396-406.
88. Angelini C, Semplicini C, Ravaglia S, Bembi B, Servidei S, Moggio M. Long-term follow-up effects on enzyme replacement treatment of adult form of acid maltase deficiency myopathy. Acta Myol 2011; 30; 152. [ Links ]
89. Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol 2013; 260: 951-9. [ Links ]
90. Anderson LJ, Henley W, Wyatt KM, et al. Effectiveness of enzyme replacement therapy in adults with late-onset Pompe disease: results from the NCS-LSD cohort study. J Inherit Metab Dis 2014; 37: 945-52. [ Links ]
91. Gungor D, Kruijshaar ME, Plug I, et al. Impact of enzyme replacement therapy on survival in adults with Pompe disease: results from a prospective international observational study. Orphanet J Rare Dis 2013;8: 49. [ Links ]
92. Schoser B, Stewart A, Kanters S, et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol 2017; 264: 621-30. [ Links ]
93. Levine JC, Kishnani PS, Chen YT, Herlong JR, Li JS. Cardiac remodeling after enzyme replacement therapy with acid alpha-glucosidase for infants with Pompe disease. Pediatr Cardiol 2008; 29: 1033-42. [ Links ]
94. Gelb MH, Turecek F, Scott CR, Chamoles NA. Direct multiplex assay of enzymes in dried blood spots by tandem mass spectrometry for the newborn screening of lysosomal storage disorders. J Inherit Metab Dis 2006; 29: 397-404. [ Links ]
95. Marsden D. Infantile onset Pompe disease: a report of physician narratives from an epidemiologic study. Genet Med 2005; 7: 147-50. [ Links ]
96. Winchester B, Bali D, Bodamer OA, et al. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab 2008;93: 275-81. [ Links ]
97. Young SP, Zhang H, Corzo D, et al. Long-term monitoring of patients with infantile-onset Pompe disease on enzyme replacement therapy using a urinary glucose tetrasaccharide biomarker. Genet Med 2009; 11: 536-41. [ Links ]
98. Taratuto AL, Dubrovsky A, Corderi J. Non-diagnostic muscle biopsies in late onset Pompe’s Disease. J Neuropathol Exp Neurol 2011; 70S: 533.
99. Chen YT, Kishnan PS, Koeberl D.Glycogen storage diseases. The online metabolic and molecular bases of inherited disease. En: https://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62672129&jumpsectionID=62672143; consultado marzo 2018. [ Links ]
100. Chien YH, Hwu WL. A review of treatment of Pompe disease in infants. Biologics 2007; 1: 195-201. [ Links ]
101. Kroos MA, Pomponio RJ, Hagemans ML, et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 2007; 68: 110-5. [ Links ]
102. Regnery C, Kornblum C, Hanisch F, et al. 36 months observational clinical study of 38 adult Pompe disease patients under alglucosidase alfa enzyme replacement therapy. J Inherit Metab Dis 2012; 35: 837-45. [ Links ]
103. Carlier RY, Laforet P, Wary C, et al. Whole-body muscle MRI in 20 patients suffering from late onset Pompe disease: Involvement patterns. Neuromuscul Disord 2011; 21: 791-9. [ Links ]
104. Gaeta M, Barca E, Ruggeri P, et al. Late-onset Pompe disease (LOPD): correlations between respiratory muscles CT and MRI features and pulmonary function. Mol Genet Metab 2013; 110: 290-6. [ Links ]
105. Gruhn KM, Heyer CM, Guttsches AK, et al. Muscle imaging data in late-onset Pompe disease reveal a correlation between the pre-existing degree of lipomatous muscle alterations and the efficacy of long-term enzyme replacement therapy. Mol Genet Metab Rep 2015; 3: 58-64. [ Links ]
106. Hoeksma M, Boon M, Niezen-Koning KE, van Overbeek-van Gils L, van Spronsen FJ. Isolated elevated serum transaminases leading to the diagnosis of asymptomatic Pompe disease. Eur J Pediatr 2007; 166: 871-4. [ Links ]
107. Kimura S. Electrodiagnosis in diseases of nerve and muscle: Principles and practice. 3rd ed, New York: Oxford University Press, 2001. [ Links ]
108. Alejaldre A, Diaz-Manera J, Ravaglia S, et al. Trunk muscle involvement in late-onset Pompe disease: study of thirty patients. Neuromuscul Disord 2012; 22: S148-S54. [ Links ]
109. Barohn RJ, McVey AL, DiMauro S. Adult acid maltase deficiency. Muscle Nerve 1993; 16: 672-6. [ Links ]
110. Manganelli F, Ruggiero L. Clinical features of Pompe disease. Acta Myol 2013; 32: 82-4. [ Links ]
111. Abresch RT, Han JJ, Carter GT. Rehabilitation management of neuromuscular disease: the role of exercise training. J Clin Neuromuscul Dis 2009; 11: 7-21. [ Links ]
112. Clarkson PM. Exercise-induced muscle damage--animal and human models. MedSciSports Exerc 1992; 24: 510-1. [ Links ]
113. Case LE, Kishnani PS. Physical therapy management of Pompe disease. Genet Med 2006; 8: 318-27. [ Links ]
114. Slonim AE, Bulone L, Goldberg T, et al. Modification of the natural history of adult-onset acid maltase deficiency by nutrition and exercise therapy. Muscle Nerve 2007; 35: 70-7. [ Links ]
115. Terzis G, Dimopoulos F, Papadimas GK, et al. Effect of aerobic and resistance exercise training on late-onset Pompe disease patients receiving enzyme replacement therapy. Mol Genet Metab 2011; 104: 279-83. [ Links ]
116. Olsen DB, Orngreen MC, Vissing J. Aerobic training improves exercise performance in facioscapulohumeral muscular dystrophy. Neurology 2005; 64: 1064-6. [ Links ]
117. van den Berg LE, Favejee MM, Wens SC, et al. Safety and efficacy of exercise training in adults with Pompe disease: evalution of endurance, muscle strength and core stability before and after a 12 week training program. Orphanet J Rare Dis 2015; 10: 87. [ Links ]
118. Orngreen MC, Olsen DB, Vissing J. Aerobic training in patients with myotonic dystrophy type 1. Ann Neurol 2005; 57: 754-7. [ Links ]
119. Sveen ML, Jeppesen TD, Hauerslev S, Kober L, Krag TO, Vissing J. Endurance training improves fitness and strength in patients with Becker muscular dystrophy. Brain 2008; 131: 2824-31. [ Links ]
120. Griffin JL. Infantile acid maltase deficiency. I. Muscle fiber destruction after lysosomal rupture. Virchows Archiv B, Cell pathology including molecular pathology 1984; 45: 23-36. [ Links ]
121. Fowler WM, Jr. Role of physical activity and exercise training in neuromuscular diseases. Am J Phys Med Rehabil. 2002; 81: S187-95. [ Links ]
122. Eagle M. Report on the muscular dystrophy campaign workshop: exercise in neuromuscular diseases Newcastle, January 2002. Neuromuscul Disord 2002; 12: 975-83. [ Links ]
123. Nilsson MI, Samjoo IA, Hettinga BP, et al. Aerobic training as an adjunctive therapy to enzyme replacement in Pompe disease. Mol Genet Metab 2012; 107: 469-79. [ Links ]
124. Cori GT. Biochemical aspects of glycogen deposition disease. Bibl Paediatr 1958; 14: 344-58. [ Links ]
125. Favejee MM, Huisstede BM, Bussmann JB, Kruijshaar ME, van der Ploeg AT. Physiotherapy management in late-onset Pompe disease: clinical practice in 88 patients. Mol Genet Metab 2012; 107: 111-5. [ Links ]
126. Bembi B, Ciana G, Martini C, et al. Efficacy of multidisciplinary approach in the treatment of two cases of nonclassical infantile glycogenosis type II. J Inherit Metab Dis 2003; 26: 675-81. [ Links ]
127. Terzis G, Krase A, Papadimas G, Papadopoulos C, Kavouras SA, Manta P. Effects of exercise training during infusion on late-onset Pompe disease patients receiving enzyme replacement therapy. Mol Genet Metab 2012; 107: 669-73. [ Links ]
128. McDonald CM. Limb contractures in progressive neuromuscular disease and the role of stretching, orthotics, and surgery. Phys Med Rehabil Clin N Am 1998; 9: 187-211. [ Links ]
129. Kinali M, Messina S, Mercuri E, et al. Management of scoliosis in Duchenne muscular dystrophy: a large 10-year retrospective study. Dev Med Child Neurol 2006; 48: 513-8. [ Links ]
130. Janicki JA, Alman B. Scoliosis: Review of diagnosis and treatment. Paediatr Child Health 2007; 12: 771-6. [ Links ]
131. Mullender M, Blom N, De Kleuver M, et al. A Dutch guideline for the treatment of scoliosis in neuromuscular disorders. Scoliosis 2008; 3: 14. [ Links ]
132. Schuller A, Wenninger S, Strigl-Pill N, Schoser B. Toward deconstructing the phenotype of late-onset Pompe disease. Am J Med Genet C Semin Med Genetics 2012; 160C: 80-8. [ Links ]
133. Case LE, Beckemeyer AA, Kishnani PS. Infantile Pompe disease on ERT: update on clinical presentation, musculoskeletal management, and exercise considerations. Am J Med Genet C Semin Med genetics 2012; 160C: 69-79. [ Links ]
134. Sarwark J, Sarwahi V. New strategies and decision making in the management of neuromuscular scoliosis. Orthop Clin North Am 2007; 38: 485-96. [ Links ]
135. van den Berg LE, Zandbergen AA, van Capelle CI, et al. Low bone mass in Pompe disease: muscular strength as a predictor of bone mineral density. Bone 2010; 47: 643-9. [ Links ]
136. Bertoldo F, Zappini F, Brigo M, et al. Prevalence of asymptomatic vertebral fractures in late-onset Pompe disease. J Clin Endocrinol Metab 2015; 100: 401-6. [ Links ]
137. Case LE, Bjartmar C, Morgan C, et al. Safety and efficacy of alternative alglucosidase alfa regimens in Pompe disease. Neuromuscul Disord 2015; 25: 321-32. [ Links ]
138. Biggar WD, Bachrach LK, Henderson RC, Kalkwarf H, Plotkin H, Wong BL. Bone health in Duchenne muscular dystrophy: a workshop report from the meeting in Cincinnati, Ohio, July 8, 2004. Neuromuscul Disord 2005; 15: 80-5. [ Links ]
139. Case LE, Hanna R, Frush DP, et al. Fractures in children with Pompe disease: a potential long-term complication. Pediatr Radiol 2007; 37: 437-45. [ Links ]
140. Papadimas GK, Terzis G, Spengos K, et al. Bone mineral density in adult patients with Pompe disease. Bone 2011; 48: 417. [ Links ]
141. Kinali M, Banks LM, Mercuri E, Manzur AY, Muntoni F. Bone mineral density in a paediatric spinal muscular atrophy population. Neuropediatrics 2004; 35: 325-8. [ Links ]
142. Papadimas G, Terzis G, Papadopoulos C, et al. Bone density in patients with late onset Pompe disease. Int J Endocrinol Metab2012; 10: 599-603. [ Links ]
143. Papadimas GK, Terzis G, Methenitis S, et al. Body composition analysis in late-onset Pompe disease. Mol Genet Metab 2011; 102: 41-3. [ Links ]
144. Jones HN, Crisp KD, Asrani P, Sloane R, Kishnani PS. Quantitative assessment of lingual strength in late-onset Pompe disease. Muscle Nerve 2015; 51: 731-5. [ Links ]
145. Sayers SP, Clarkson PM, Rouzier PA, Kamen G. Adverse events associated with eccentric exercise protocols: six case studies. Med Sci Sports Exerc 1999; 31: 1697-702. [ Links ]
146. Hobson-Webb LD, Jones HN, Kishnani PS. Oropharyngeal dysphagia may occur in late-onset Pompe disease, implicating bulbar muscle involvement. Neuromuscul Disord 2013; 23: 319-23. [ Links ]
147. Chan J, Desai AK, Kazi ZB, et al. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol Genet Metab 2017; 120: 163-72. [ Links ]
148. Bernstein DL, Bialer MG, Mehta L, Desnick RJ. Pompe disease: dramatic improvement in gastrointestinal function following enzyme replacement therapy. A report of three later-onset patients. Mol Genet Metab 2010; 101: 130-3. [ Links ]
149. Ravaglia S, Pichiecchio A, Rossi M, et al. Dietary treatment in adult-onset type II glycogenosis. J Inher Metabol Dis 2006; 29: 590. [ Links ]
150. Slonim AE, Bulone L, Minikes J, et al. Benign course of glycogen storage disease type IIb in two brothers: nature or nurture? Muscle Nerve 2006; 33: 571-4. [ Links ]
151. Schoser B, Hill V, Raben N. Therapeutic approaches in glycogen storage disease type II/Pompe Disease. Neurotherapeutics 2008; 5: 569-8. [ Links ]
152. Kobayashi H, Shimada Y, Ikegami M, et al. Prognostic factors for the late onset Pompe disease with enzyme replacement therapy: from our experience of 4 cases including an autopsy case. Mol Genet Metab 2010; 100: 14-9. [ Links ]
153. Ravaglia S, Danesino C, Moglia A, et al. Changes in nutritional status and body composition during enzyme replacement therapy in adult-onset type II glycogenosis. Eur J Neurol 2010; 17: 957-62. [ Links ]
154. Kishnani PS, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr 2006; 149: 89-97. [ Links ]
155. Meikle PJ, Grasby DJ, Dean CJ, et al. Newborn screening for lysosomal storage disorders. Mol Genet Metab 2006; 88: 307-14. [ Links ]
156. Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid alpha.-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 2007; 68: 99-109. [ Links ]
157. Geel TM, McLaughlin PM, de Leij LF, Ruiters MH, Niezen-Koning KE. Pompe disease: current state of treatment modalities and animal models. Mol Genet Metab 2007; 92: 299-307. [ Links ]
158. Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet 2000; 356: 397-8. [ Links ]
159. van Til NP, Stok M, Aerts Kaya FS, et al. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood 2010; 115: 5329-37. [ Links ]
160. Van den Hout JM, Kamphoven JH, Winkel LP, et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 2004; 113: e448-57. [ Links ]
161. Rossi M, Parenti G, Della Casa R, et al. Long-term enzyme replacement therapy for pompe disease with recombinant human alpha-glucosidase derived from chinese hamster ovary cells. J Child Neurol 2007; 22: 565-73. [ Links ]
162. Orlikowski D, Pellegrini N, Prigent H, et al. Recombinant human acid alpha-glucosidase (rhAGA) in adult patients with severe respiratory failure due to Pompe disease. Neuromuscul Disord 2011; 21: 477-82. [ Links ]
163. Gungor D, Kruijshaar ME, Plug I, et al. Quality of life and participation in daily life of adults with Pompe disease receiving enzyme replacement therapy: 10 years of international follow-up. J Inher Metabol Dis 2016; 39: 253-60. [ Links ]
164. Laloui K, Wary C, Carlier RY, Hogrel JY, Caillaud C, Laforet P. Making diagnosis of Pompe disease at a presymptomatic stage: to treat or not to treat? Neurology 2011; 77: 594-5. [ Links ]
165. Spada M, Porta F, Vercelli L, et al. Screening for later-onset Pompe’s disease in patients with paucisymptomatic hyperCKemia. Mol Genet Metab 2013; 109: 171-3.
166. Wagner M, Chaouch A, Muller JS, et al. Presymptomatic late-onset Pompe disease identified by the dried blood spot test. Neuromuscul Disord 2013; 23: 89-92. [ Links ]
167. Kwon JM, Steiner RD. “I’m fine; I’m just waiting for my disease”: the new and growing class of presymptomatic patients. Neurology 2011; 77: 522-3.
168. Echaniz-Laguna A, Carlier RY, Laloui K, et al. Should patients with asymptomatic pompe disease be treated? A nationwide study in France. Muscle Nerve 2015; 51: 884-9. [ Links ]
169. Al Jasmi F, Al Jumah M, Alqarni F, et al. Diagnosis and treatment of late-onset Pompe disease in the Middle East and North Africa region: consensus recommendations from an expert group. BMC Neurol 2015; 15: 205. [ Links ]
170. Raben N, Jatkar T, Lee A, et al. Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol Ther 2002; 6: 601-8. [ Links ]
171. Raben N, Danon M, Gilbert AL, et al. Enzyme replacement therapy in the mouse model of Pompe disease Mol Genet Metab 2003; 80: 159-69. [ Links ]
172. Masat E, Laforet P, De Antonio M, et al. Long-term exposure to Myozyme results in a decrease of anti-drug antibodies in late-onset Pompe disease patients. Sci Rep 2016; 6: 36182. [ Links ]
173. Rohman PJ, Scott E, Richfield L, Ramaswami U, Hughes DA. Pregnancy and associated events in women receiving enzyme replacement therapy for late-onset glycogen storage disease type II (Pompe disease). J Obstet Gynaecol Res 2016; 42: 1263-71. [ Links ]
174. Perniconi B, Vauthier-Brouzes D, Morelot-Panzini C, et al. Multidisciplinary care allowing uneventful vaginal delivery in a woman with Pompe disease. Neuromuscul Disord 2016; 26: 610-3. [ Links ]
175. O’Brien D. Aids to the examination of the peripheral nervous system. Edinburgh: Saunders-Elsevier, 2010.
176. ATS statement: guidelines for the six-minute walk test. ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. Am J Respi Crit Care Med 2002; 166: 111-7. [ Links ]
177. McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve 2013; 48: 343-56. [ Links ]
178. Hernández-Vidal P, Berrios GE, Bulbena A. Concepto y evaluación de la sensación de fatiga. En: Bulbena A, Berrios GE, Fernández de Larrinoa P, eds. Medición clínica en psiquiatría y psicología. Barcelona: Masson, 2000, p 123-35. [ Links ]
179. Merkies IS, Schmitz PI, Van Der Meche FG, Samijn JP, Van Doorn PA. Psychometric evaluation of a new handicap scale in immune-mediated polyneuropathies. Muscle Nerve 2002; 25: 370-7. [ Links ]
180. Hagemans ML, Laforet P, Hop WJ, et al. Impact of late-onset Pompe disease on participation in daily life activities: evaluation of the Rotterdam Handicap Scale. Neuromuscul Disord 2007; 17: 537-43. [ Links ]
181. Ware JE Jr., Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care 1992; 30: 473-83. [ Links ]

