











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkPUNTOS CLAVE
Conocimiento actual
• El descubrimiento de un nuevo principio activo compren de el estudio sobre los procesos de obtención, caracteri zación de la actividad y estudios preclínicos para evaluar su toxicidad. El uso de biomarcadores bioquímicos como criterios de valoración sustitutos, se encuentra en auge para diferentes modelos experimentales.
Contribución del artículo al conocimiento actual
• En esta revisión, se evalúa la validación apropiada de técnicas empleadas para el dosaje de biomarcadores bioquímicos en estudios preclínicos, bajo normas BPL, y se describen los inconvenientes asociados al bajo volumen de muestra disponible, y al cumplimiento de las normas vigentes supeditado a las realidades de los laboratorios analíticos.
El descubrimiento de un nuevo principio activo com prende el estudio de los procesos de síntesis, el aislamien to desde una fuente natural o la obtención biotecnológica, la caracterización de su actividad y la fase preclínica completa, donde se incluye la evaluación toxicológica. Estos estudios tienen como objetivo demostrar que el compuesto es aceptable, en cuanto a su eficacia y segu ridad, para un ensayo en seres humanos en las diferentes fases clínicas. Se ha descripto que solo 1 de cada 10 000 moléculas descubiertas, con posible actividad terapéuti ca, pasa a la fase de desarrollo farmacéutico, 1 de cada 100 000 supera los ensayos clínicos y logra registrarse, y solo 3 de cada 10 nuevos medicamentos registrados recuperan su inversión inicial1,2. Por otra parte, en los casos de medicamentos oncológicos que superan exitosamente las etapas preclínicas y clínicas y llegan al registro, pueden tener un recupero de las inversiones cercano a 55 dólares por cada dólar invertido3. Por este motivo, el diseño racional de fármacos y de métodos para su estudio constituye una herramienta indispensable en el desarrollo actual de nuevos medicamentos, contribu yendo al aumento de las posibilidades de éxito y a una disminución de los costos.
Evaluación de fármacos y dispositivos biomédicos. Estudios preclínicos
En la actualidad, se descubren o sintetizan cada año más de 1000 nuevos productos entre sustancias quími cas, medicamentos, cosméticos, plaguicidas, aditivos alimentarios, etc. Al respecto, y de acuerdo al avance de la investigación biológica en medicina humana y veteri naria, resulta indiscutible la importancia que han tenido los modelos animales. Las sustancias mencionadas anteriormente, ya sea en las etapas de investigación básica o en las preclínicas, deben estudiarse mediante el desarrollo de protocolos experimentales adecuados que, cualquiera sea su naturaleza, implican la interacción de reactivos físicos, químicos y/o biológicos.
Sumado a esto, los cambios continuos en las tecno logías de dispositivos médicos plantean desafíos únicos en la evaluación de la biocompatibilidad de los productos. Por ejemplo, la valoración de los productos polimerizados in situ requiere especial atención en la selección y el di seño de los ensayos adecuados para evaluar la toxicidad potencial de los monómeros y otras sustancias químicas involucradas en el proceso de polimerización. Estos dis positivos requieren estudios especiales de implantación para documentar el curso completo de la degradación y reabsorción del mismo, a fin de demostrar que el sitio del implante no se altera de manera significativa y que la degradación no produce efectos tóxicos. Para la correcta evaluación de los resultados, las presentaciones ante los organismos de control deben incluir una justificación de la elección de los ensayos realizados y la validación de todos los métodos y técnicas aplicadas, siguiendo están dares basados en normas internacionales4-7. Además, es esencial que todos los datos, incluyendo los resultados de las pruebas realizadas, estén plenamente documen tados, bajo sistemas de buenas prácticas de laboratorio auditables8.
La primera parte del estudio de nuevos fármacos consiste en la realización de pruebas físicas y químicas orientadas a determinar principalmente la susceptibilidad a la degradación de moléculas potencialmente útiles. Usualmente las moléculas más inestables son rápida mente descartadas o se modifican químicamente para aumentar su estabilidad. Posteriormente, las moléculas más estables son evaluadas en su actividad biológica en diversos modelos experimentales, incluyendo culti vos celulares, órganos aislados, ensayos en animales de experimentación, etc. El uso de modelos animales presupone que los principios fisiológicos y patológicos básicos son comunes en diferentes especies, sobre todo si comparten características genéticas. Este supuesto rara vez se cumple debiendo realizarse pruebas en más de una especie, como lo determina la autoridad regulatoria de cada país9.
Las pruebas biológicas son los primeros ensayos que se realizan para comprobar tanto la eficacia como la segu ridad de un nuevo fármaco y pueden llegar a condicionar la continuidad del estudio y el desarrollo. La prueba de eficacia no solo implica la observación del efecto propia mente dicho, sino un estudio toxicocinético, farmacoci nético y farmacodinámico. Las pruebas de seguridad en esta fase deben contemplar la determinación global de la toxicidad (aguda, subaguda y crónica), los posibles efectos sobre el sistema reproductor y la embriogénesis, y la posibilidad de mutagénesis y/o carcinogénesis. En estas pruebas se usan dosis muy superiores a aquellas que se supone serán usadas definitivamente en la clínica, lo que permite la detección de respuestas tóxicas de baja frecuencia. Todos estos procedimientos están compren didos dentro de la fase preclínica del estudio de nuevos fármacos, debido a que pueden conducir a la prueba del nuevo fármaco en humanos, etapa que se conoce como fase clínica. En la fase preclínica es crucial la selección adecuada del modelo experimental, ya que al costo del ensayo se le suma el de la síntesis del nuevo producto que en general es muy elevado. Esto ha llevado a la ne cesidad de utilizar animales de baja masa corporal (como los ratones, de 25 gramos aproximadamente), donde se reduce significativamente el volumen de las sustancias a administrar y consecuentemente se reduce el volumen de la muestra de sangre a obtener en cada punto de mues treo para la determinación de biomarcadores asociados a la toxicidad10.
El uso de biomarcadores como sustitutos de los resultados en ensayos clínicos, deben cumplir con de terminados criterios, como por ejemplo: ser confiables de acuerdo a la recopilación de datos de seguridad ade cuados; demostrar una clara asociación entre el resultado subrogado (biomarcador) y la capacidad para predecir beneficios significativos en los pacientes; estudios alea torizados de drogas de la misma clase que demuestren que cambios en el sustituto conduce a mejorías clínicas relevantes; evidencia de efectos precisos y duraderos al usar los sustitutos y beneficios que superen los daños, entre otros aspectos11.
Definiciones de biomarcador
Diferentes publicaciones han descripto la aplicación de biomarcadores en el desarrollo de fármacos, utilizando diversas nomenclaturas para describir aspectos distintos de este proceso4,5,12. La estandarización de la terminolo gía para facilitar la comprensión de la literatura conduce a la definición consensuada de que un biomarcador es un indicador que mide y evalúa objetivamente los procesos biológicos normales, procesos patológicos o respuestas farmacológicas a una intervención terapéutica. El punto final clínico se define como una variable que mide cómo los pacientes se sienten, funcionan o sobreviven, mientras que el punto final sustituto, es un biomarcador que está destinado a sustituir el punto final clínico. En este caso, se espera que un parámetro sustituto, prediga el beneficio clínico. Otra distinción crítica que debe hacerse cuando un biomarcador experimenta una validación del método analítico, es compararlo con una calificación clínica.
El uso de biomarcadores como criterios de valoración sustitutos en un ensayo clínico, requiere la unificación de los criterios de valoración clínicos que se están sustitu yendo, la clase de intervención terapéutica que se aplica y las características de la población y el estado de la enfermedad en el que se realiza la sustitución. El término sustituto significa literalmente “sustituir”; por lo tanto, se desaconseja el uso del término marcador sustituto porque el término sugiere que la sustitución es por un marcador, más que por un criterio de valoración clínico11.
La validación del método analítico es el proceso de evaluación del ensayo, sus características de rendimiento y las condiciones óptimas que generarán la reproduci bilidad y exactitud del ensayo. La calificación clínica es el proceso probatorio de vincular un biomarcador con procesos biológicos y puntos finales clínicos. Mientras que la “validación” y la “calificación” o “evaluación” se han utilizado indistintamente en la literatura, la distinción debe hacerse para describir adecuadamente la fase particular que el biomarcador está atravesando en el proceso de desarrollo de fármacos. Como tal, el término “validación” está reservado para los métodos analíticos, y la “califi cación” para la evaluación clínica de los biomarcadores. Tanto los procesos de validación como los de calificación están entrelazados y, por lo tanto, su integración guía el desarrollo de biomarcadores con el principio de vincular un biomarcador con su uso previsto5.
Importancia de los biomarcadores en ensayos preclínicos
En los últimos años, los biomarcadores clínicos integran parte del diagnóstico y seguimiento de numerosas en fermedades. En este sentido, un biomarcador se define como una sustancia elaborada por un tejido específico que puede ser detectada en la circulación sanguínea. Para ser útil desde el punto de vista clínico, debe liberarse en una cantidad proporcional a la evolución del proceso patológico concreto y proporcionar información sobre la presencia, gravedad y el pronóstico de la enfermedad12. Lo ideal sería que el biomarcador fuera estable y fácil de detectar mediante análisis disponibles, rápidos y económicos. Sin embargo, esto no sucede en el caso de animales de laboratorio, debido a que los diseños de estudios se basan generalmente en la medicina humana y en las principales especies de interés veterinario. Los biomarcadores se utilizan en medicina habitualmente para evaluar la función de diferentes órganos. El número de biomarcadores con potencial interés clínico crece cada año, y muchos de ellos han sido aprobados por agencias reguladoras o están en fases avanzadas de investiga ción. Clásicamente, por ejemplo, la urea y la creatinina son biomarcadores para controlar la función renal, y la bilirrubina total, fosfatasa alcalina (FAS), alanina amino transferasa (ALT), aspartato aminotransferasa (AST) y gamma glutamil transferasa (GGT) lo son para evaluar la lesión hepatocelular o hepatobiliar, la amilasa para evaluar la integridad pancreática, creatina quinasa (CK) y lactato deshidrogenasa (LDH) para el tejido muscular, FAS para metabolismo óseo, incluyendo también metabolitos (glu cosa, colesterol, triglicéridos, proteínas totales, albúmina) e iones (Ca, P, K), entre muchos otros5,3.
Un biomarcador debe ser un parámetro que pueda ser objetivamente medido y evaluado como indicador de un proceso biológico normal o patológico, o como respuesta a una intervención terapéutica. Los biomarcadores far macodinámicos se refieren específicamente a aquellos asociados a la duración de una respuesta farmacológica, mientas que los biomarcadores de punto final son aquellos que permiten predecir el beneficio clínico o la ausencia de él durante el estudio de un nuevo fármaco4.
Los biomarcadores son ampliamente utilizados en el desarrollo de nuevos fármacos, contribuyendo a incre mentar la productividad y mejorando el cuidado de los pacientes a través de numerosas acciones durante la fase traslacional del desarrollo de un medicamento. Reciente mente ha aumentado el estudio de biomarcadores como consecuencia de su extensa aplicación en el desarrollo preclínico y clínico de un fármaco. Este hecho permite acelerar el proceso de registro y aprobación, aumentar la eficiencia y facilitar la selección de la dosis antes de los ensayos clínicos de fase III, que son los de mayor costo para la industria farmacéutica14. Los biomarcadores utiliza dos en los ensayos de toxicidad y eficacia pueden ayudar a identificar riesgos en los modelos animales y mejorar los futuros ensayos clínicos, contribuyendo a la selección adecuada de dosis y a la estratificación de pacientes. Es por ello que los biomarcadores adecuados contribuyen a descartar tempranamente fármacos potencialmente tóxicos, aumentando la productividad en las etapas de desarrollo y reduciendo el tiempo y los costos asociados. Debido al gran aporte de los biomarcadores en la industria farmacéutica y de diagnóstico se han convertido en foco de nuevas e importantes validaciones y regulaciones15.
Los biomarcadores han sido incluidos en la “agenda de trabajo” de la National Institute of Health (NIH-USA)16, con el objetivo de facilitar una investigación médica más efectiva y productiva. Además, la Federal Drug Admi nistration (FDA-USA), ha generado una iniciativa para identificar las fuentes que generan las mayores demo ras y otros problemas durante el desarrollo de nuevos fármacos, citando específicamente la importancia de contar con herramientas adecuadas para la evaluación preclínica entre las que se mencionan los biomarcadores bioquímicos6.
Como ya hemos mencionado, los avances en las cien cias básicas asociadas al descubrimiento y desarrollo de fármacos, ha llevado a un incremento significativo en el número de potenciales candidatos terapéuticos, sin em bargo, esto no se traduce linealmente en un incremento de medicamentos aprobados para su uso14. Consecuen temente, los esfuerzos para mejorar la eficiencia de este proceso están siendo implementados permanentemente en el ámbito de la investigación traslacional15. En este sentido, el uso de biomarcadores constituye una herra mienta fundamental para mejorar la eficiencia del proceso de desarrollo de nuevos medicamentos.
Para comprender la importancia de los biomarcadores en estudios preclínicos, a continuación, se enumeran los biomarcadores más importantes en relación a diferentes órganos y sistemas:
Biomarcadores hepáticos
El hígado es un órgano crítico debido a su amplio papel en la homeostasis y el metabolismo, la desintoxicación y excreción de xenobióticos y compuestos endógenos. Ejemplos de la función hepática son la síntesis de albúmina y los factores de coagulación, la síntesis de novo y almacenamiento de glucosa, el uso y reciclaje de bilirrubina y colesterol, el metabolismo de hormonas y la degradación de diferentes medicamentos17.
La lesión hepática inducida por fármacos se encuentra entre las principales causas por las cuales éstos han sido retirados del mercado en las últimas tres décadas18,19. Es así que se ha ido incrementado el alcance y la complejidad de los biomarcadores en estudios preclínicos, con el fin de lograr mejorar la predicción de una posible lesión hepática generada por ciertas drogas. Varios marcadores tradicio nales indican lesión hepática tanto en especies clínicas como en las preclínicas. Sin embargo, los conceptos de lesión hepática y función hepática deben diferenciarse cuidadosamente, para lograr comprender y diferenciar las respuestas tóxicas del hígado a las diferentes lesiones19,20. Es importante recordar que puede existir una alteración biológica estadísticamente significativa en los biomarca dores de lesión hepática, pero sin evidenciar efecto algu no en los índices de función hepática, y viceversa. Con respecto a lesión hepática, a su vez podemos diferenciar entre lesión hepatocelular y hepatobiliar17.
La recomendación actual para analizar la seguridad en estudios preclínicos, sugiere usar un mínimo de cuatro parámetros séricos para evaluar la lesión hepatocelular y hepatobiliar18. La actividad de ALT, AST, la sorbitol deshidrogenasa (SDH) y/o la actividad de la glutamato deshidrogenasa (GLDH) evaluarán la lesión hepatocelu lar, mientras que la actividad de FAS, la GGT, la bilirru bina total, los ácidos biliares totales evaluarán la lesión hepatobiliar. Entre estos parámetros séricos, ALT, AST, FAS y la bilirrubina total también se usan para la identifi cación clínica de lesiones hepáticas en humanos, por lo que tienden a usarse con mayor frecuencia (Tabla 1). Es importante tener en cuenta que la elección y utilidad de estos biomarcadores varía entre las diferentes especies involucradas en estudios preclínicos17. Entre los biomarca dores tradicionales de lesión hepatocelular, se encuentra la ALT, la cual se la considera un indicador sensible y traducible de esta lesión en especies preclínicas comunes. La actividad de la ALT sérica es el indicador de los efectos hepatotóxicos generados por fármacos21, aunque no siem pre se correlaciona bien con los datos histopatológicos obtenidos22. Debido a la falta de especificidad de la ALT para definir una lesión hepatocelular, a menudo se utilizan pruebas complementarias de química clínica e histopa tología para ayudar a interpretar los valores obtenidos. Una regla útil para la interpretación de los valores de ALT como signo de lesión hepática, en ausencia de resultados histopatológicos, es la regla de los dos biomarcadores. La misma considera que una elevación en tres veces los valores normales de ALT, combinado con una alteración significativa en otro biomarcador de lesión hepática, es un signo de lesión hepatocelular23. Después de la introduc ción de un nuevo fármaco en estudios preclínicos a corto plazo, especialmente en ausencia de otros indicadores de lesión hepática, la respuesta de ALT se produce como elevaciones transitorias, las cuales no son consideradas clínicamente relevantes, mientras que los valores vuelvan a la normalidad después de una exposición continua al fármaco24. La magnitud del incremento de ALT suele ser mayor que el de AST cuando ambas enzimas están ele vadas debido a una lesión hepatocelular. Esto se debe a que la ALT tiene una vida media más larga que la AST y que la mayor fracción de AST se une a las mitocondrias25.

La actividad de la GLDH aumenta con la lesión he patocelular y sus valores basales pueden ser mayores y más duraderos que la ALT26. La GLDH es una enzima sérica clásica como la ALT y AST. A diferencia de la ALT, la actividad de la GLDH sérica no se ve afectada por el daño del músculo esquelético ni inducida por los corticos teroides. Si bien la GLDH no se usa de manera rutinaria en estudios preclínicos, debido a la baja disponibilidad de reactivos confiables, está dejando de ser un biomarcador clásico de poco uso, por la ventaja para detectar lesión hepática aguda en comparación con la ALT17.
La sorbitol deshidrogenasa (SDH) es un indicador sensible y específico de una lesión hepatocelular aguda en ratones, perros, primates y también en humanos22. Sin embargo, la SDH tiene una vida media corta (4 horas en caninos) y una estabilidad limitada en comparación con la mayoría de otras enzimas importantes en la química clínica, por lo que se requiere que se realice su determi nación inmediatamente luego de la toma de la muestra23. Hasta el momento, se desconoce si en ciertos estudios, la cuantificación de la SDH solo aporta un valor adicional junto a la cuantificación de la ALT17.
Los ácidos biliares totales son importantes en el meta bolismo hepático del colesterol y la absorción de grasas y vitaminas liposolubles. Los niveles de ácidos biliares circulantes están influenciados por la dieta y el ayuno, pero el aumento en sus valores también puede deberse a un cambio funcional en el hígado27. Su cuantificación es un complemento útil para evaluar la extensión y las con secuencias de la lesión hepática, ya que se lo considera un índice de la función hepática en especies preclínicas.
La FAS es un biomarcador principal de lesión hepa tobiliar en especies preclínicas comunes. Al igual que la ALT, la FAS no es específica para la lesión hepatobiliar, ya que se puede observar un aumento de la actividad de esta enzima en condiciones de crecimiento y/o enferme dad ósea28. Las fluctuaciones de FAS deben interpretarse con cautela en ensayos preclínicos, porque hay una iso forma intestinal de la FAS que provoca una disminución de la actividad de la misma, particularmente en ratones, durante el ayuno o la anorexia, y un aumento transitorio posprandial19.
La bilirrubina total es un producto de la degradación de la hemoglobina de los glóbulos rojos envejecidos. Las elevaciones de este biomarcador suelen ser indicativas de lesión hepatobiliar o alteración de la función hepática29.
La GGT se eleva por inducción en condiciones de le sión hepatobiliar y puede usarse para confirmar las eleva ciones de la actividad de FAS asociada a esta patología19.
La actividad sérica de la malato deshidrogenasa (MDH) se la correlaciona con la lesión hepática, aunque una lesión cardíaca puede provocar también una elevación de la actividad de esta enzima. Al igual que la GLDH, los niveles de MDH son estables en pacientes sanos de todos los sexos y grupos de edad, pero están elevados a causa de una lesión hepática30.
Por su parte, la cuantificación de proteínas totales (PT), las cuales en su mayoría se sintetizan en el híga do, excepto las gammaglobulinas, son de sumo interés, ya que su dosaje puede indicar si existe un problema hepático, renal, intestinal, hemorrágico o inflamatorio. La concentración de PT se determina generalmente en suero o plasma31. La albúmina se sintetiza sólo en el hígado y una disminución en su concentración sérica se denomina hipoalbuminemia, la cual, si es significativa, puede deberse a una lesión hepática. No se registra incrementos en la síntesis de albúmina relacionado con afecciones hepáticas. El hígado es el único órgano que convierte el amoniaco en urea. En una disfunción hepática grave, pero también en una dieta pobre en proteínas, la concentración sérica de urea está disminuida31.
Es importante también destacar el dosaje de otros biomarcadores esenciales para evaluar el correcto fun cionamiento del hígado, como son la cuantificación sérica de colesterol, triglicéridos y glucosa. El colesterol es el precursor de los ácidos biliares, que son importantes para la digestión y absorción de lípidos de la dieta en el intes tino. Si el valor del colesterol total está aumentado, es un indicador de lesión hepatobiliar. Por su parte, valores altos de triglicéridos pueden indicar una obstrucción biliar y en una hepatitis crónica puede observarse una disminución de este biomarcador. La glucosa es la principal fuente de energía de los tejidos y su concentración en sangre está controlada por la insulina, el glucagón, la adrenalina y el cortisol. La glucosa no es un buen analito para evaluar la función hepática, ni para hacer un diagnóstico hepático, ya que solo en los casos más severos de una insuficien cia hepática se observa una hipoglucemia significativa31.
Biomarcadores renales
La nefrotoxicidad y la lesión renal aguda son de ocurrencia común en los estudios preclínicos de seguridad y conti núa siendo una de las principales razones del descarte de nuevos fármacos y/o prometedores candidatos. Los riñones son especialmente susceptibles a una amplia gama de xenobióticos debido a que reciben una gran pro porción del volumen sanguíneo circulante (hasta el 25% del volumen de sangre bombeada por el corazón), lo que da como resultado la exposición a una elevada cantidad de compuestos y/o sus metabolitos. Las nefronas partici pan en el proceso de formación de la orina, que además concentran las sustancias tóxicas en el líquido tubular renal. Entre las nefrotoxinas más importantes se incluyen metales pesados como el cromo, plomo y el mercurio, agentes terapéuticos antiinflamatorios no esteroideos como el acetaminofeno, antibióticos aminoglucósidos como por ejemplo la gentamicina, y agentes inmunosu presores y quimioterapéuticos como la ciclosporina o el cisplatino32. Los clásicos biomarcadores que se valoran como indicadores de la función renal, son la valoración de la urea, expresada como nitrógeno ureico en sangre (BUN), la creatinina sérica y el fósforo. La creatinina se elimina con más facilidad y generalmente se incrementa después que la urea, por eso, para evaluar la función renal, es importante determinar en el suero las concentra ciones de urea y creatinina31. La creatinina sérica es una prueba imprecisa, ya que no refleja con precisión la tasa de filtración glomerular cuando la función renal cambia constantemente33.
Debido al importante papel de los riñones en el mante nimiento de la homeostasis del fósforo, una concentración elevada de este catión en suero sugiere una enfermedad renal crónica avanzada que puede conducir a problemas de salud sustanciales, principalmente enfermedades óseas y cardiovasculares34. Sin embargo, estos paráme tros son indicadores relativamente insensibles de lesión renal y no demuestran alteraciones discernibles hasta que ocurra una falla funcional de hasta un 65-75% de la capacidad renal35.
Claramente, el rendimiento de estos biomarcadores no es suficiente para controlar la lesión renal en muchos ensayos preclínicos. La necesidad de obtener indicadores renales con mayor sensibilidad, ha llevado al descubri miento de una serie de biomarcadores de nueva gene ración que proporcionan una detección más temprana y más específica de la lesión renal para su uso en el desarrollo de fármacos (Tabla 2). Inicialmente, la FDA calificó un conjunto de biomarcadores urinarios en ratas para controlar la lesión renal inducida por fármacos junto con el dosaje de la urea (BUN) y la creatinina sérica36.

Entre estos biomarcadores se encuentran la albúmina, las proteínas totales, la cistatina C, la clusterina, la molécula de lesión renal 1 y la β2-microglobulina17.
Es importante señalar que la calificación de un biomar cador, por parte de un organismo regulador, representa la aprobación de su uso en circunstancias muy específicas, en una especie determinada, y no descarta la utilidad ni impide la inclusión de datos de otros biomarcadores con fines informativos con el fin de proporcionar una perspecti va adicional. Los biomarcadores renales generalmente se clasifican en varios grupos que indican su asociación con la lesión de una parte específica de la nefrona, como por ejemplo el túbulo proximal, glomérulo o el conducto colec tor. Sin embargo, un trabajo reciente sugiere que existe una buena cantidad de superposición y redundancia entre los biomarcadores individuales con respecto al segmento específico de la nefrona, por lo tanto, clasificar los efectos de biomarcadores en el glomérulo o los túbulos parece ser el enfoque más apropiado en la mayoría de los casos37.
Biomarcadores de lesiones musculares cardíacas y esqueléticas
La cardiotoxicidad puede ser causada por una variedad de xenobióticos y terapias químicas que incluyen agentes antiarrítmicos como los bloqueadores adrenérgicos β, compuestos inotrópicos positivos como la digoxina o el isoproterenol, anestésicos como el isoflurano o el propofol, narcóticos como la cocaína y agentes antineoplásicos como la doxorrubicina. Es importante distinguir entre los biomarcadores asociados a efectos cardiotóxicos a aquellos biomarcadores vinculados con efectos sobre el sistema cardiovascular. Los biomarcadores utilizados para la identificación y caracterización de los efectos de la lesión cardíaca, se pueden clasificar en términos generales como aquellos que identifican la lesión de los miocitos cardíacos estructurales, como las troponinas y las proteínas de unión a ácidos grasos y aquellos que indican alteraciones en la función cardíaca como los péptidos natriuréticos. Aunque la lesión cardíaca se puede detectar a través de una variedad de métodos que inclu yen electrocardiogramas y otro tipo de evaluaciones, la cuantificación de biomarcadores en sangre y otros fluidos corporales (suero o plasma), son los más utilizados en estudios preclínicos y en estudios de toxicología17.
Los dos biomarcadores más importantes para la detección de la lesión o la necrosis aguda de miocitos cardíacos incluyen la medición de la troponina I cardía ca (cTnI) y/o la troponina T (cTnT) en suero o plasma. Ambas moléculas están involucradas en la regulación del proceso de excitación y acoplamiento del aparato contráctil dentro del miocito cardíaco, y se liberan a la circulación luego de una lesión celular o necrosis. Estos biomarcadores han demostrado repetidamente su alta sensibilidad y especificidad en la detección de lesiones agudas del miocardio en una variedad de enfermedades y condiciones experimentales tanto en humanos como en ratas38,39. Las elevaciones en estos biomarcadores a menudo precederán a la primera evidencia histológica de lesión cardíaca y se correlacionarán con los efectos observados mediante ecocardiografía y con las puntua ciones generales de daño histológico40,41. En animales sanos, las troponinas cardíacas circulan en niveles bajos, casi indetectables, y pueden aumentar en cuestión de horas después de una lesión40. La detección de estos biomarcadores a nivel plasmático (señal positiva) es de corta duración ya que el clearance de las moléculas de cTnI es rápido y ocurre dentro de 24 a 48 horas después de la primera lesión. Dada la sensibilidad de la mayoría de los ensayos comerciales para determinar troponina, generalmente se estima necesario observar un aumento de más del doble en los valores de cTnI antes de consi derar que la señal es biológicamente o toxicológicamente significativa42. Al igual que con la mayoría de los ensayos de biomarcadores más nuevos, la falta de métodos estan darizados hace que la comparación de valores absolutos entre diferentes técnicas y laboratorios sea problemática. Se debe considerar cuidadosamente el método de dosaje y los rangos históricos específicos del laboratorio antes de determinar la interpretación de las señales positivas. Existen moléculas de troponina similares que también están presentes en el músculo esquelético como la troponina esquelética (sTnI), pero son antigénicamente distintas de sus contrapartes cardíacas, lo que aumenta la especificidad de ambos grupos de biomarcadores para la detección de lesiones musculares esqueléticas y cardíacas, respectivamente43. Aunque cTnI y cTnT tienen una utilidad interpretativa y características cinéticas simi lares, los ensayos de cTnI han sido mejor caracterizados, están ampliamente disponibles y han demostrado una buena reactividad cruzada y capacidad de traducción entre múltiples especies. La cTnI es el biomarcador más utilizado para la detección de lesiones miocárdicas en las evaluaciones de seguridad farmacológica39.
Históricamente, se han utilizado diferentes biomar cadores enzimáticos para la detección de lesiones de miocitos cardíacos en humanos y animales (Tabla 3). Estos incluyen a la AST, la LDH, la CK y la isoenzima creatina quinasa MB (CK-MB), que ya no se recomien dan para su uso como biomarcadores cardíacos debido a su baja especificidad, su rendimiento inconsistente entre especies, su amplia distribución en los tejidos y su falta de relevancia clínica44. Solo sirven para identificar señales inflamatorias generales y siempre deben usarse junto con otros biomarcadores cardíacos más específicos. Sin embargo, muchos de estos biomarcadores son muy adecuados como biomarcadores de la lesión del músculo esquelético.

La toxicidad o la lesión del músculo esquelético son un hallazgo común en los estudios preclínicos, luego de la exposición de los animales a diferentes compuestos quí micos como por ejemplo los inhibidores de la colinesterasa o el carbofurano. A diferencia de la extensa investigación dedicada al desarrollo de ensayos para el diagnóstico de la enfermedad miocárdica, se ha dedicado muy poco esfuerzo al desarrollo de biomarcadores para la detección de lesiones del músculo esquelético. Tradicionalmente, se han utilizado una serie de enzimas como la CK, AST o la LDH y diferentes biomarcadores de proteínas como por ejemplo la mioglobina, pero generalmente estos ca recen de la sensibilidad y especificidad necesaria para distinguir de manera confiable los efectos sutiles en el músculo esquelético de los efectos ocasionados en otros tejidos. La CK es una enzima citosólica que se encuentra en una gran variedad de tejidos, incluidos el músculo esquelético y cardíaco, el cerebro, en menor medida el tracto gastrointestinal, el útero, la vejiga y el riñón45. Es el biomarcador más utilizado para detectar lesiones musculares generales y tiene tres isoenzimas distintas (CK-MM, CK-BB y CK-MB), que pueden determinarse mediante separación electroforética. Tradicionalmente, CK-MM y CK-MB se han utilizado como biomarcadores de lesión muscular esquelética y cardíaca, respectiva mente; sin embargo, la mayor parte de la actividad de CK en el músculo cardíaco y esquelético se atribuye a la CK-MM44. También existe un alto grado de variabilidad en el contenido de CK y las proporciones de sus isoenzi mas entre ubicaciones musculares específicas, tipos de músculos (contracción rápida/contracción lenta) y entre especies, particularmente en lo que respecta a la actividad miocárdica de CK-MB46,47. La CK es un buen biomarcador para la lesión del músculo esquelético, pero la isoenzima CK-MB carece de la especificidad y consistencia entre especies para ser utilizada sola como un biomarcador cardíaco específico.
La LDH es una enzima que se encuentra en la mayoría de las células, incluidas células musculares esqueléticas y cardíacas, hepatocitos y eritrocitos, con una distribución muy variable entre los diferentes tipos de tejidos y entre especies35,48. Dada su amplia distribución en los tejidos, el aumento en la actividad de la LDH no es específico de un solo órgano. La actividad de la LDH es general mente mayor en el músculo esquelético y el hígado en comparación con el músculo cardíaco, lo que respalda su uso como biomarcador de una lesión en estos dos tipos de músculos48. Se han realizado intentos de usar algunas isoenzimas de la LDH como la LDH1 o la LDH2, para aumentar la especificidad en el músculo cardíaco, obteniendo resultados similares a los de las isoenzimas de la CK, ya que existe una considerable variabilidad entre los diferentes tipos de músculos, entre especies y concentraciones dentro del corazón que lo excluye como un biomarcador útil de lesión cardíaca44.
La AST y ALT son enzimas que se encuentran, entre otros, en el músculo esquelético e hígado. El aumento de las actividades de AST generalmente se asocia con efectos sobre el hígado y el músculo esquelético, pero también se observa con afecciones hemolíticas35. El aumento de ALT se asocia clásicamente con una lesión hepatocelular; sin embargo, los casos de lesión muscular significativa también se han asociado con aumentos en la actividad de esta enzima49. Recientemente, se han des cubierto otros biomarcadores con una mejor especificidad para el músculo esquelético y están ganando una mayor aceptación. El más prometedor de estos es la sTnI, se ha demostrado que es un biomarcador sensible y específico para la lesión del músculo esquelético50.
La sTnI es similar, pero antigénicamente distinta de su contraparte cardíaca específica, la cTnI, y se ha demos trado que ofrece la capacidad de distinguir entre el daño esquelético y cardíaco en ratas donde se han probado una variedad de diferentes toxinas cardíacas y miotoxi nas51. Este biomarcador también se ha incluido en varios paneles de lesiones musculares destinados a diferenciar entre la lesión muscular cardíaca y esquelética.
Control y validación de técnicas analíticas
Las normativas más utilizadas para el control y validación de técnicas analíticas son la guía de la Agencia Europea de Medicamentos (EMA: European Medicines Agency)7, la guía de la FDA-USA6 y las normas de la Conferencia internacional de armonización (ICH: International Con ference of Armonization)52. Estas guías definen los ele mentos claves y las cifras de mérito a evaluar, necesarias para la validación de las técnicas analíticas destinadas a medir las concentraciones de biomarcadores en matrices biológicas como suero, plasma u orina; obtenidas en estudios toxicocinéticos en animales y en todas las fases de ensayos clínicos.
Los criterios sugeridos por las guías FDA y la EMA para la validación de un método bioanalítico son similares, pero no idénticos, ya que las cifras de méritos requeridas en ambas guías son diferentes. Las fortalezas de la guía de la FDA recaen en la facilidad de su interpretación ya que se presenta mucha información en los anexos. Por otro lado, la guía EMA describe la conducta práctica de los experimentos con mayor precisión, en comparación a la FDA53.
Los parámetros a determinar para la validación de las técnicas analíticas se describen a continuación (las diferencias entre las distintas guías son aclaradas)
Linealidad
Se define como la capacidad o la habilidad de producir resultados que son directamente proporcionales a la concentración del analito, dentro de un rango dado52. Los requerimientos que debe cumplir la curva de calibrado para asegurar su linealidad, de acuerdo a las guías son:
- Se deben ejecutar 6 estándares de calibración, al menos por duplicado y se deben espaciar de manera uniforme. El coeficiente de determinación (R2) para la curva estándar debe ser ≥ 0.956,7.
- El coeficiente de variación porcentual (CV%) de los replicados de los estándares debe ser ≤ 20%; 25% en el límite inferior de cuantificación (LLOQ: lower limit of cuantification) y en el límite superior de cuantificación (ULOQ: upper limit of cuantification). En el caso de tratarse de triplicados, y de no cumplir con este criterio, puede descartarse el valor de absorbancia más alejado, considerándolo como error técnico6,7.
- La concentración de cada punto de la curva, cal culados nuevamente en base a la ecuación de la recta obtenida, debe ser 100 ± 20% (25% en los extremos) con respecto a la concentración nominal para al menos el 75% de los estándares de calibración que se encuentren dentro del rango cuantificable de la curva6,7.
- La concentración más baja en la curva de calibración debe ser el LLOQ, si se cumplen las condiciones descrip tas en los ítems anteriores6,7.
- El estándar más alto definirá al ULOQ de la meto dología analítica6,7.
La unión de química pura y aplicada (IUPAC: The Union of Pure and Applied Chemistry) desalienta los co eficientes de correlación como indicadores de linealidad en la relación entre concentración y señal. Para probar la linealidad, sugiere a los autores que deben informar el valor experimental de F correspondiente a la relación de la varianza residual al error puro al cuadrado, y la F crítica tabulada para la comparación. Esta prueba es el in dicador de linealidad más adecuado y equivale a verificar estadísticamente si la varianza residual es mayor que el error puro al cuadrado derivado del estudio de muestras replicadas (método de regresión de los cuadrados míni mos). Se sugiere cumplir, además de los requerimientos descriptos por las guías anteriormente enunciadas, con el método de regresión de los cuadros mínimos, ya que existen ejemplos donde la correlación es cercana a 1 pero no cumplen con este método54.
Rango
El rango de un procedimiento analítico se define como el intervalo entre la concentración superior e inferior (defi nidas por el LLOQ y el ULOQ) de analito en la muestra (incluidas estas concentraciones) para el cual el procedi miento analítico ha demostrado tener un nivel adecuado de precisión, exactitud y linealidad52.
Precisión y exactitud
La precisión se define como un procedimiento analítico que expresa la cercanía (grado de dispersión) entre una serie de mediciones obtenidas del muestreo múltiple de la misma muestra homogénea en las condiciones pres critas. Puede considerarse en tres niveles: repetibilidad, precisión intermedia y reproducibilidad52.
- Repetibilidad (precisión intra-ensayo): es la precisión obtenida bajo las mismas condiciones de operación en un intervalo corto de tiempo (mismo día), por un mismo analista, en la misma muestra homogénea y en el mismo equipo.
- Precisión intermedia (precisión inter-ensayo): es la precisión obtenida bajo las mismas condiciones de operación en tres días distintos por el mismo o diferen tes analistas, en la misma muestra homogénea y en el mismo equipo.
- La reproducibilidad expresa la precisión entre labora torios (en el caso de estudios colaborativos, generalmente aplicados a la estandarización de la metodología).
La precisión debe investigarse utilizando muestras homogéneas. Sin embargo, si no es posible obtenerla, puede analizarse utilizando muestras preparadas artifi cialmente o una solución de muestra. La precisión de un procedimiento analítico generalmente se expresa como la varianza, la desviación estándar o el coeficiente de variación porcentual (CV%) de una serie de mediciones52.
Para la FDA, la precisión determinada en cada nivel de concentración no debe exceder el 15% del CV%, excepto para el LLOQ, donde no debe exceder el 20% del CV%6. En cambio, para EMA, la precisión determinada en cada nivel de concentración no debe exceder el 20% del CV%, excepto para el LLOQ y el ULOQ, donde no debe exceder el 25% del CV%7.
La exactitud de un procedimiento analítico expresa la cercanía entre el valor que se acepta como uno verdadero convencional o de referencia aceptado y el valor encontra do. También se la conoce como veracidad52. La exactitud de un método analítico describe la cercanía de la media de los resultados de la prueba obtenidos por el método utilizado con el valor real (concentración) del analito. Se determina mediante el análisis replicado de muestras que contienen cantidades conocidas del analito (es decir, controles internos). La exactitud debe medirse utilizando un mínimo de cinco determinaciones por concentración. Se recomienda un mínimo de tres controles internos en el rango de concentraciones de muestra de estudio es peradas. Para la FDA, el valor medio obtenido debe estar comprendido entre el 85 y el 115% del valor nominal, ex cepto en el LLOQ, donde debe estar comprendido entre el 80 y el 120%. La desviación de la media del valor nominal sirve como medida de exactitud6. En cambio, para EMA, el valor medio obtenido debe estar comprendido entre el 80 y el 120% del valor nominal, excepto en el LLOQ y en el ULOQ, donde este debe estar entre el 75 y el 125% del valor nominal7.
Límite de detección y de cuantificación
El límite de detección (LOD: limit of detection), se define como la mínima concentración que da una señal estadís ticamente distinta del blanco, y el límite de cuantificación (LOQ: limit of cuantification), se define como la menor cantidad de analito que puede ser determinado en la muestra con valores aceptables de precisión y exactitud, bajo las condiciones experimentales establecidas52.
El LOD generalmente se estima utilizando la defini ción anterior, ahora abandonada por IUPAC, basada en la concentración de analito que proporciona una señal al menos tres veces mayor que la desviación estándar de la señal del blanco. Este valor de LOD suele ser una subestimación55,56. La recomendación moderna de IUPAC primero requiere definir un nivel para el límite de detección (LD), lo que implica un cierto riesgo de falsas detecciones (también llamados falsos positivos, errores α o errores de tipo I). Como se ilustra en la Figura 1, el área sombreada en verde representa una parte de la distribución gaussiana de los valores de concentración que tienen una probabi lidad de declarar a un analito “analito ausente”, mientras que en realidad está presente. Este es el significado de la detección falsa o el falso positivo. El LOD se define como un nivel de concentración para el cual el riesgo de una falsa detección (falsos negativos, errores β o errores de Tipo II) tengan una probabilidad β. Esto corresponde al área sombreada en rojo en la Figura 1, donde el analito puede declararse presente mientras de hecho está au sente. A los errores α y β, generalmente se les asignan valores razonablemente pequeños, dependiendo de la aplicación analítica específica.
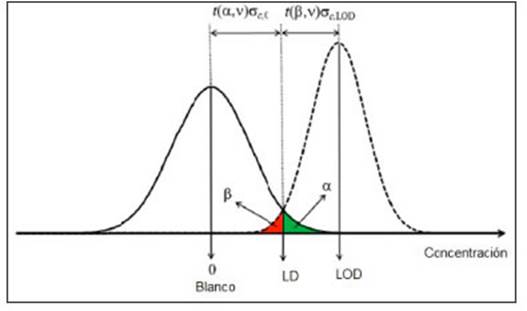
Fig. 1 Ilustración de la definición oficial (IUPAC) del límite de decisión (LD) y el límite de detección (LOD). Dos bandas gaussianas se centran en el blanco y en el LOD, respectivamente. El LD ayuda a decidir si el analito se detecta o no con una tasa a de falsas detecciones, mientras que el LOD implica la detección con una tasa a de falsas detecciones y una tasa b de falsas no detecciones. Las áreas sombreadas corresponden a la tasa de falsas detecciones (verde) y falsas no detecciones (rojo)36
La fórmula entonces para calcular el LOD es la si guiente:

donde t(α,v) y t(β,v) son coeficientes de Student con n gra dos de libertad y probabilidades α y β, respectivamente, σc,0 y σc,LOD son los errores estándar de concentración para los niveles en blanco y LOD, A es la pendiente de la curva de calibrado, I es el número de muestras de calibración, Sx / y es la desviación estándar residual y h0 viene dado por la siguiente ecuación:

donde ccal es la concentración de la calibración media y ci es cada uno de los valores de concentración de la calibración.
Conceptos similares se aplican al LOQ, que se calcula mediante la fórmula:

donde el factor 10 asegura una incertidumbre relativa máxima del 10%.
Cabe señalar que la Comisión Europea ha adoptado recomendaciones similares57, incluida la capacidad de detección (CCβ), que es la concentración más pequeña de la sustancia que se puede detectar, identificar y/o cuantificar en una muestra con una probabilidad de error de β. Esto puede interpretarse como la concentración mínima de analito que puede discriminarse del blanco, controlando los riesgos de falsos positivos y falsos negati vos. La definición de CCβ es análoga a la LOD anterior51.
Selectividad
La selectividad se define como la capacidad de medir el analito de interés en presencia de compuestos no relacio nados en la matriz. La selectividad se prueba agregando al menos 10 fuentes de interferencias como por ejemplo muestras lipémicas o hemolizadas en el LLOQ o cerca de él. La selectividad debe evaluarse en el extremo inferior de un ensayo donde ocurren problemas en la mayoría de los casos. También puede ser prudente evaluar la selectividad a concentraciones de analitos más altas. En los casos en que la interferencia depende de la concen tración, es esencial determinar la concentración mínima donde se produce la interferencia. Puede ser necesario ajustar el nivel más bajo de cuantificación en consecuen cia, antes de la validación del ensayo. La exactitud debe estar comprendida entre el 80 y el 120% (75 al 125% en el LLOQ) de la concentración nominal enriquecida en al menos el 80% de las matrices evaluadas7.
Linealidad dilucional
En casos donde el rango de la curva de calibración es muy estrecho, es necesario demostrar con muestras de estándares de calibración que el analito de interés, cuando está presente en concentraciones que exceden el rango de cuantificación (por encima del ULOQ), puede medirse con precisión después de diluirlo en una matriz blanco, para llevar las concentraciones del analito dentro del ran go validado para el análisis. La concentración calculada para cada dilución debe estar comprendida entre el 80 y el 120% de la concentración nominal, después de la corrección por la dilución realizada, y la precisión de las concentraciones finales en todas las diluciones no debe exceder el 20% del CV%7.
Efecto matriz
El efecto matriz (ME: matrix effect) se define como la al teración o interferencia directa o indirecta en la respuesta debido a la presencia de analitos no deseados u otras sustancias interferentes en la muestra52.
El ME se evalúa mediante el método de adición de estándar54. Describe la desviación entre la pendiente de las curvas de calibración matriciales y la pendiente de las curvas de calibración estándar externas, utilizando la siguiente ecuación:

El resultado de % ME = cero, significa que no se ha observado señal, supresión o mejora; % ME ± 10, sig nifica un efecto matriz nulo; % ME ± (10 - 20) significa un efecto matriz suave; % ME ± (20 - 50), significa un efecto matriz medio y un % ME + 50, significa un efecto matriz fuerte58.
Estabilidad
Los estudios de estabilidad se efectúan para determinar el período de tiempo y las condiciones de almacena miento en las cuales el analito se mantiene dentro de las especificaciones sobre identidad, potencia, calidad y pureza52.
Las muestras para evaluar estabilidad se deben com parar con los calibradores y/o los controles de calidad re cién preparados. Se deben evaluar al menos tres réplicas en cada una de las concentraciones, bajas y altas. Las evaluaciones de la estabilidad del analito deben realizarse en la misma matriz que las muestras del estudio. Todas las determinaciones de estabilidad deben usar muestras preparadas a partir de una solución madre recién pre parada. Las condiciones usadas en los experimentos de estabilidad deben reflejar las situaciones que proba blemente se encuentren durante el manejo y el análisis de la muestra real (por ejemplo, almacenamiento a largo plazo, a temperatura ambiente, y ciclos de congelación-descongelación). También debe evaluarse la estabilidad de la solución madre. Para la FDA, los resultados de las muestras de estabilidad deben estar comprendidos entre el 85 y el 115% de las concentraciones nominales6. Para la EMA, los resultados de las muestras de estabilidad deben estar comprendidos entre el 80 y el 120% de las concentraciones nominales7.
Problemática en la determinación de biomarcadores endógenos
Como en muchos otros campos de análisis, demostrar la validez de los métodos bioanalíticos se ha vuelto cada vez más relevante. En los últimos 15 años, la validación de un método bioanalítico ha sido objeto de arduo debate, y han surgido varios documentos, directrices y publica ciones acerca de esta temática59,60. Como resultado, los principios de validación para estos métodos se encuentran ahora razonablemente bien establecidos. El enfoque general es que los parámetros de validación importantes como por ejemplo la selectividad, precisión, exactitud y estabilidad se determinan usando muestras enriquecidas y que se utilizan como modelo para las llamadas mues tras incurridas (es decir, muestras tomadas de sujetos humanos o animales después de la administración de un compuesto en particular). El supuesto subyacente es, por lo tanto, que están disponibles tanto una matriz libre de analito como un estándar de referencia del analito bien caracterizado. La determinación cuantitativa de los compuestos endógenos, es decir, naturales en muestras biológicas, es más complicada, tanto analíticamente como desde un punto de vista de la validación. A menudo es difícil, sino imposible, obtener muestras sin analitos de la matriz biológica auténtica o muestras con concentra ciones de analitos conocidas con precisión, por lo que la preparación de muestras de referencia debe abordarse de una manera diferente y, como consecuencia, la validación también se vuelve menos sencilla. Los compuestos en dógenos se han medido en laboratorios clínicos durante muchos años y con una variedad de técnicas, pero el interés en la determinación de estos biomarcadores, como parte del proceso de desarrollo de nuevos fármacos, es relativamente reciente. Como se dijo anteriormente, las decisiones críticas sobre la continuación de un programa de desarrollo de fármacos se basan frecuentemente en concentraciones de uno o más biomarcadores bien seleccionados, ya que estos proporcionan información crucial sobre la eficacia y la seguridad de un fármaco. La validación de las técnicas analíticas para analitos endógenos, hasta ahora se ha visto obstaculizada por la ausencia de directrices oficiales. Muchos investigadores intentan aplicar los principios de validación de métodos para ensayos de fármacos, en particular los emitidos por la FDA, también a sus métodos de analitos endógenos, a fin de garantizar resultados con un nivel de calidad comparable, pero cabe señalar que estos principios no estaban destinados a compuestos endógenos y, en mu chos casos, no pueden aplicarse directamente61.
Una parte esencial del desarrollo del método es la se lección de una forma adecuada de preparar muestras de calibración y control de calidad. Idealmente, estas mues tras son partes (alícuotas) de la matriz biológica auténtica que contiene una concentración conocida exacta del ana lito auténtico. Para compuestos endógenos, la matriz bio lógica auténtica típicamente contiene una concentración desconocida del analito, lo que lo hace inadecuado para la preparación de muestras de referencia. Un enfoque bien conocido para solucionar este problema es la aplicación del método de adición estándar. Al agregar concentra ciones crecientes del analito a alícuotas individuales de la muestra de interés, se crea una curva de calibración y la concentración endógena en la muestra se determina a partir de la intersección de esta curva de calibración con el eje de las abscisas. Aunque este enfoque se ha aplicado ampliamente y durante muchos años, y ciertamente tiene aspectos atractivos (por ejemplo, descarta los efectos de la matriz), generalmente requiere un mayor volumen de muestra (en el caso de ratones el volumen de muestra es limitante) y tiempo de análisis de lo que puede permitirse en el bioanálisis contemporáneo61.
La FDA propone que para aquellos analitos que también sean compuestos endógenos, la precisión de la medición de éstos es un desafío cuando el ensayo no puede distinguir entre el agente terapéutico y la contra parte endógena. En tales situaciones, recomienda los siguientes enfoques para validar y controlar el rendimiento del ensayo:
- La matriz biológica utilizada para preparar los están dares de calibración debe ser la misma que las muestras de estudio y estar libre del analito endógeno. Para abor dar la idoneidad del uso de una matriz biológica libre de analitos, se debe demostrar que la matriz tiene: a) ningún analito endógeno medible; y b) sin ME o interferencia cuando se compara con la matriz biológica.
- El uso de matrices alternativas debe estar justificado. Los controles deben prepararse añadiendo cantidades conocidas del analito en la misma matriz biológica que las muestras de estudio.
- Las concentraciones endógenas del analito en la matriz biológica deben evaluarse mediante análisis repe tido de las mismas. Las concentraciones de los controles deben tener en cuenta las concentraciones endógenas en la matriz biológica (es decir, aditivo) y ser representativas de las concentraciones esperadas del estudio6.
Estado actual de la tecnología
La realización de ensayos preclínicos demanda una tecno logía acorde a los requisitos de las entidades regulatorias y a la complejidad de las respuestas requeridas por los patrocinantes. En la clínica, se han utilizado los biomarca dores bioquímicos para monitorear el proceso terapéutico, la progresión de las enfermedades y la eficiencia de las intervenciones médicas. Recientemente los mismos han sido aplicados al desarrollo de fármacos, aunque, como se mencionó previamente, las tecnologías no se han adaptado a las necesidades específicas de los modelos de animales utilizados en la investigación traslacional. La principal limitante es la falta de técnicas analíticas vali dadas que permitan la realización de un panel complejo de biomarcadores con el escaso volumen de muestra obtenible del ratón, por ejemplo, la principal especie utilizada para ensayos preclínicos a nivel mundial y en la cual se han desarrollado numerosos modelos de enfer medades humanas, a través de modificaciones genéticas específicas. Sumado a esto, toda tecnología desarrollada requiere, para su uso en estudios preclínicos de registro, ser validada de acuerdo a los más altos estándares de las agencias regulatorias internacionales4,15,61.
Dentro de estos estándares internacionales, las Buenas Prácticas de Laboratorio (BPL) son un concepto de gestión que abarca los procesos organizativos y las condiciones en las cuales los estudios de laboratorio se planifican, aplican, monitorean, registran e informan. Es tos principios aplican a entidades de ensayo que llevan a cabo estudios de seguridad preclínicos con fines de regis tro de productos farmacéuticos, cosméticos, pesticidas, fármacos de uso veterinario, aditivos para la alimentación humana y animal, y productos químicos, con objeto de obtener datos acerca de sus propiedades y/o seguridad para ser presentados a las autoridades regulatorias competentes. Su cumplimiento ha sido considerado una herramienta fundamental para el desarrollo en el sector estratégico SALUD del PAI2020. Las BPL constituyen un estándar aceptado y promovido por la Organización para la Cooperación y el Desarrollo Económico (OCDE) que proveen datos confiables y de alta calidad en el desarrollo de estudios preclínicos de seguridad de los productos mencionados previamente con fines de registro. A partir de septiembre de 2011, Argentina, a través del Organismo Argentino de Acreditación (OAA), en su carácter de Auto ridad Nacional de Monitoreo de las BPL de la OCDE, ha alcanzado su condición de Adherente Pleno al Acuerdo de Aceptación Mutua de Datos (MAD) para la Evaluación de Químicos de la OCDE. Dicha adhesión, permite que los datos generados por las entidades de ensayo ins peccionadas por el OAA, y declaradas en conformidad con las BPL, sean aceptados por los países miembros y adherentes plenos, y de este modo, minimizar las barreras no tarifarias al comercio. De este modo, Argentina avanza hacia un marco de armonización global que fortalece la integración económica, eliminando potenciales barreras no comerciales entre los países miembros y no miembros de la OCDE. La calidad de los datos es un problema que posee una importante dimensión internacional. Si las autoridades normativas de un país pueden poner su con fianza en los datos procedentes de otro país, la repetición tanto de las pruebas como de los gastos incurridos, se podrá evitar a los gobiernos, así como a las industrias. Por otra parte, los principios de BPL comunes facilitan el intercambio de información e impiden la aparición de obs táculos no arancelarios para los intercambios, al mismo tiempo que contribuyen para la protección de la salud y del medio ambiente8.
Conclusiones
En esta revisión se ha documentado que el uso de bio marcadores constituye una herramienta fundamental para mejorar la eficiencia del proceso de desarrollo de nuevos medicamentos. Se presentaron varios biomarcadores utilizados de forma rutinaria para evaluar diferentes teji dos y sistemas de órganos. Una característica clave de la interpretación de los datos de estos biomarcadores es tener en cuenta la heterogeneidad de las señales positi vas de respuesta en el tiempo, magnitud y por especie, y lograr una buena comprensión del rendimiento analítico de cada ensayo individual. Un análisis cuidadoso, caso por caso, e integrador de los datos de estos biomarcadores, proporciona una mejor perspectiva sobre los riesgos po tenciales de los problemas de seguridad e informa sobre las estrategias apropiadas de mitigación de riesgos en el desarrollo de fármacos. El objetivo final es minimizar los costos del desarrollo y, dependiendo de los resultados obtenidos, se puede llegar a condicionar la continuidad del estudio.
Por otro lado, en base a los conocimientos actuales, no se cuentan comercialmente con las técnicas analíticas necesarias para realizar estas determinaciones en peque ños volúmenes de muestra, considerando que la especie más utilizada en las etapas preclínicas es el ratón, por su baja masa corporal y en la cual se han desarrollado numerosos modelos de enfermedades humanas.
La determinación cuantitativa de biomarcadores en dógenos, en muestras biológicas, presenta una serie de complicaciones para su validación y para el cumplimiento de los requerimientos detallados en las normas vigentes. Por lo tanto, el desafío que se presenta está centrado en el desarrollo y en la validación de técnicas analíticas es pecíficas para la determinación de los biomarcadores de interés contando con muy bajos volúmenes de muestra, y el amoldamiento de las normas vigentes a las realidades y las posibilidades de los laboratorios que las realizan.

