











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkPUNTOS CLAVE
• El síndrome de Guillain-Barré clásico (SGB) es una neuropatía sensitivo-motora ascendente de inicio agudo, que puede presentarse de forma atípica o como variante clínica.
• Los resultados anormales en estudios electrofisiológicos y una combinación de aumento del nivel de proteínas con recuento normal de células (disociación albúmino-citológica) en el líquido cefalorraquídeo son caracterís ticas clásicas del SGB, pero puede haber resultados normales en ambas pruebas, especialmente al comienzo de la enfermedad.
• Es necesario supervisar la función respiratoria en todos los pacientes, ya que puede producirse insuficiencia respiratoria sin síntomas de disnea.
• La inmunoglobulina intravenosa y la plasmaféresis son igualmente eficaces en el tratamiento del SGB; ningún otro tratamiento ha demostrado ser eficaz.
• Si bien la eficacia del tratamiento repetido, en pacientes que mostraron respuesta clínica insuficiente, es incierta, es práctica común en los que muestren deterioro des pués de una respuesta inicial al tratamiento.
• La mejoría clínica suele ser más extensa a un año del inicio de la enfermedad y puede continuar durante más de cinco años.
El síndrome de Guillain-Barré (SGB) es una enfer medad inflamatoria del sistema nervioso periférico y es la causa más común de parálisis flácida aguda, con una incidencia global anual de aproximadamente 1-2 casos por cada 100 000 personas por año1. El SGB ocurre con más frecuencia en hombres que en mujeres y la inciden cia aumenta con la edad, si bien todos los grupos etarios pueden verse afectados1. Los pacientes con SGB se pre sentan típicamente con debilidad y síntomas sensitivos en las piernas que progresan hacia los brazos y los músculos cervicales y faciales, aunque la presentación clínica de la enfermedad es heterogénea y existen diversas variantes clínicas diferentes. El diagnóstico de SGB se basa en los antecedentes del paciente y en los exámenes neurológico, electrofisiológico y de líquido cefalorraquídeo (LCR)2-4. Deben descartarse otras enfermedades que producen un cuadro clínico similar al SGB4. Los estudios electrofisio lógicos proporcionan evidencia de disfunción del sistema nervioso periférico y pueden distinguir entre los subtipos de SGB: polirradiculoneuropatía desmielinizante inflama toria aguda (AIDP por sus siglas en inglés), neuropatía axonal motora aguda (AMAN por sus siglas en inglés) y neuropatía axonal sensitivo-motora aguda (AMSAN por sus siglas en inglés)5. La progresión de la enfermedad puede ser rápida y la mayoría de los pacientes alcanzan su máxima discapacidad en un plazo de dos semanas. Aproximadamente el 20% de los enfermos desarrollan falla respiratoria y requieren ventilación mecánica. Es posible que presenten arritmias cardíacas e inestabilidad de la presión arterial debido a la afectación del sistema nervioso autónomo6. Esta afectación del sistema nervioso autónomo contribuye a la mortalidad, que se estima entre el 3 y el 10% de los casos, incluso con la mejor atención médica disponible7-9. Después de la fase progresiva ini cial, se alcanza una fase de meseta que puede durar de días a semanas o meses, después de lo cual comienza la recuperación. Entre el 60 y el 80% de los pacientes pueden caminar de forma independiente 6 meses después del inicio de la enfermedad, con o sin tratamiento10,11. El SGB es una enfermedad monofásica, aunque algunos enfermos pueden presentar deterioro posterior a la mejoría o estabilización clínica con el tratamiento. Este fenómeno se conoce como fluctuación relacionada con el tratamiento. Las recaídas de SGB pueden ocurrir en entre el 2 y el 5% de los casos10,12-15.
Se cree que el SGB es ocasionado por una respuesta inmune anormal a las infecciones, lo que provoca daños en los nervios periféricos, aunque la patogénesis aún no está completamente entendida. En un subgrupo de pacientes con SGB, se encuentran anticuerpos séricos contra los gangliósidos, que residen en altas densidades en el axolema y otros componentes de los nervios peri féricos16,17. La activación del complemento, la infiltración por macrófagos y el edema son características típicas de los nervios periféricos y las raíces nerviosas afectados en pacientes con SGB16.
La incidencia de SGB puede aumentar durante los brotes de enfermedades infecciosas que desencadenan la enfermedad18. Recientemente, las epidemias del virus del Zika en la Polinesia Francesa en 2013 y en América Latina y el Caribe en 2015-2016 estuvieron relacionadas con un aumento de casos diagnosticados con SGB19-21.
La epidemia por virus del Zika sacó a la luz la falta de guías aplicables a nivel mundial para el diagnóstico y tratamiento del SGB. Estas guías resultan necesarias ya que el diagnóstico de SGB puede ser difícil debido a la heterogeneidad en la presentación clínica, un diagnóstico diferencial extenso y la falta de herramientas de diagnós tico o biomarcadores altamente sensibles y específicos. También se necesitan guías para el tratamiento y el cuidado de los pacientes con SGB, porque la progresión de la enfermedad puede variar mucho, lo que complica un abordaje totalmente prescriptivo del manejo clínico. Además, las opciones de tratamiento son limitadas y costosas, y muchos pacientes experimentan una discapa cidad residual y quejas que pueden ser de difícil manejo.
La disponibilidad de guías clínicas para el SGB, aplica bles a nivel mundial, es especialmente importante, ya que es probable que en el futuro ocurran nuevas epidemias de patógenos que lo desencadenen. Para generar esta guía aplicable a nivel mundial para el SGB, un grupo de expertos internacionales identificaron los diez pasos más importantes para su manejo, que abarcan el diagnóstico, el tratamiento, el seguimiento del paciente, el pronóstico y el control a largo plazo (Fig. 1). Para cada paso, se proporcionaron recomendaciones basadas en la eviden cia de la literatura y/o la opinión de expertos. Se buscó el consenso para cada recomendación a fin de finalizar la guía. Estas recomendaciones pretenden ayudar a los proveedores de atención médica en la toma de decisiones clínicas; sin embargo, el uso de la información de este artículo es voluntario. Los autores no asumen ninguna responsabilidad por cualquier lesión o daño a personas o propiedad que surja o esté relacionado con cualquier uso de esta información, o por cualquier error u omisión.
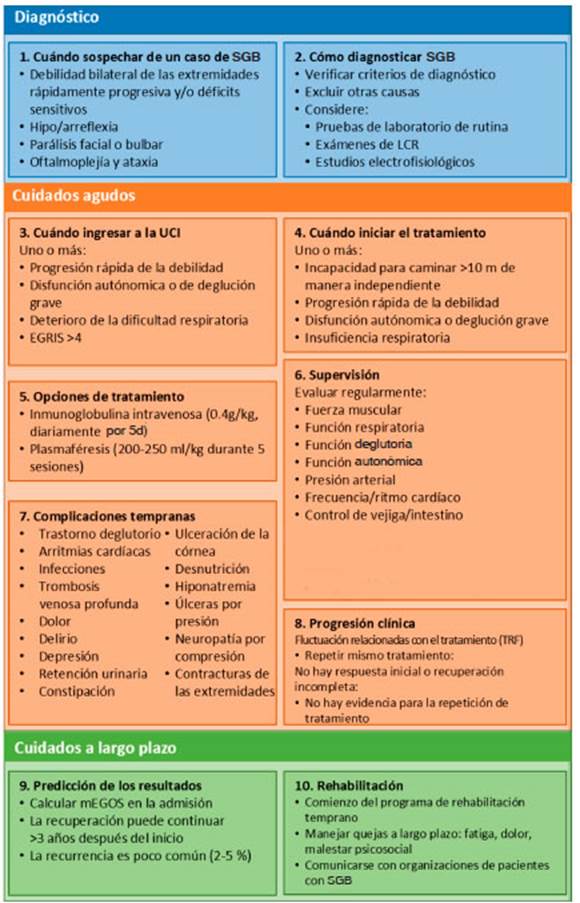
Fig. 1 Enfoque de diez pasos para el diagnóstico y el manejo del síndrome de Guillain-Barré (SGB). Este resumen de viñetas proporciona una descripción general de cada uno de los diez pasos que se describen en la guía. La frecuencia del seguimiento depende del cuadro clínico y debe evaluarse en cada paciente
Métodos
Luego de la epidemia por el virus del Zika y su asociación con un aumento en la incidencia de SGB, se conformó la Red Latinoamericana de preparación para el Zika (Zika PLAN), financiada por la Unión Europea22. Los participan tes de la red ZikaPLAN, entre los que se incluyen expertos sobre SGB de los Países Bajos (SEL, MRM y BCJ), Brasil (FG y MED) y el Reino Unido (HJW) prepararon inicial mente la nueva guía a seguir. Estos miembros aportaron su experiencia específica en clínica y en investigación a partir de sus roles de liderazgo en grandes proyectos internacionales sobre SGB (como el International GBS Outcome Study, IGOS), así como su experiencia directa en el control de grandes aumentos en los casos de SGB en regiones afectadas por el virus Zika en América La tina23. Para desarrollar los lineamientos preliminares, se llevó a cabo una serie de reuniones personales entre los autores principales del comité de redacción (SEL, MRM, BCJ y HJW), junto con reuniones individuales más pe queñas con colegas en América Latina (SEL, FG y MED) y correspondencia por correo electrónico continua para revisar borradores y recibir contribuciones. Sobre la base de la opinión de expertos y a través del consenso, este grupo identificó diez de los pasos más importantes en el diagnóstico y tratamiento del SGB.
Para cada paso, los miembros del comité de redacción (SEL y MRM) realizaron búsquedas estructuradas de lite ratura en octubre de 2018, utilizando PubMed y Embase, y los resultados de estas búsquedas proporcionaron la base para el primer borrador de esta guía. El principal criterio de inclusión para las búsquedas bibliográficas fue cualquier estudio, ensayo, revisión o reporte de caso publicado a partir de 2015 que proporcionara detalles sobre el diag nóstico, tratamiento, manejo o pronóstico de los pacientes con SGB. Se excluyeron de la revisión las publicaciones sobre la patogénesis del SGB, o aquellas centradas en enfermedades no relacionadas con el SGB, junto con las publicaciones escritas en un idioma que no fuera inglés u holandés. Las palabras clave utilizadas en la estrategia de búsqueda incluyeron los siguientes términos de Medical Subject Headings (MeSH): “Síndrome de Guillain-Barré” y [“diagnóstico” O “terapéutica” O “resultado del trata miento” O “pronóstico”]. Para obtener bibliografía sobre temas más específicos, se combinaron términos MeSH adicionales con palabras clave de búsqueda primarias, incluidas “inmunoglobulinas intravenosas”, “plasmafé resis”, “unidades de cuidados intensivos”, “embarazo”, “síndrome de Miller Fisher” y “VIH”. Después de esta re visión de la literatura más reciente, el comité de redacción (SEL, MRM, BCJ y HJW) identificó estudios destacados publicados antes de 2015 para su inclusión, junto con artículos adicionales seleccionados mediante la revisión de las referencias de los manuscritos ya incluidos y la consulta con los autores. Dentro de lo posible, nuestras recomendaciones con respecto al tratamiento se basaron en revisiones sistemáticas. Se buscó la opinión experta de los autores para obtener recomendaciones cuando se disponía de evidencia más limitada (por ejemplo, estudios de cohortes o estudios de control de casos), por ejemplo, sobre temas relacionados con el diagnóstico diferencial o la rehabilitación del SGB.
Teniendo en cuenta los diferentes modelos de aten ción en salud y las variantes clínicas del SGB, este pri mer borrador fue revisado posteriormente por un grupo internacional de expertos en SGB de Argentina (RR), Australia (EMY), Bangladesh (BI), Brasil (MLBF y CS), China (YW), Colombia (CAP), Japón (SK), Malasia (NS), Países Bajos (PA v D), Singapur (TU), Sudáfrica (KB), EE. UU. (DRC y JJS) y Reino Unido (RACH). En total, se realizaron siete rondas de revisión para llegar a un consenso. Para considerar la perspectiva de los pacientes con SGB sobre el manejo de la enfermedad, la GBS/CIDP, Fundación International, https://www.gbs-cidp.org/, una organización sin fines de lucro que brinda apoyo, educación, financiamiento de investiga ción, y protección a pacientes con SGB o polineuropatía desmielinizante inflamatoria crónica (CIDP por sus siglas en inglés) y sus familias, revisó el manuscrito y propor cionó comentarios durante el desarrollo de la guía. Esta guía fue originalmente publicada en inglés en Nature Reviews Neurology24. Para mejorar la utilidad global de estos lineamientos, y con el permiso de los editores de Nature Reviews Neurology, los hemos traducido al español. Con el fin de garantizar una traducción precisa, hemos reunido a un grupo de coautores para coordinar el proceso de traducción y revisar las traducciones. Este grupo estaba compuesto por los hablantes nativos RR, CAP, y DRC, RAH, HJW, BCJ y SEL. La traducción fue realizada por la agencia de traducción certificada ISO Etymax. El manuscrito se tradujo primero al español y luego se hizo una traducción inversa de esta traducción al inglés por un traductor independiente. El comité de revisión revisó la traducción al español y la traducción inversa al inglés y las editó si lo consideró necesario.
Paso 1: ¿Cuándo sospechar de un caso de síndrome de Guillian-Barré?
Características clínicas típicas
El SGB debe considerarse como diagnóstico en pacientes con debilidad bilateral rápidamente progresiva de piernas y/o brazos, en ausencia de afectación del sistema nervioso central u otras causas evidentes. Los pacientes con la for ma sensitivo-motora clásica de SGB presentan parestesias distales o pérdida sensitiva, acompañadas o seguidas de debilidad que comienza en las piernas y progresa hacia los brazos y los músculos craneales. Los reflejos están disminuidos o ausentes en la mayoría de los pacientes al momento de la presentación y en casi todos los pacientes en el nadir10,25. La disautonomía es común y puede incluir inestabilidad de presión arterial o de la frecuencia cardíaca, disfunción pupilar y disfunción intestinal o vesical26. El dolor es una queja frecuente y puede ser muscular, radicular o neuropático27. El inicio de la enfermedad es agudo o subagudo y los pacientes suelen alcanzar la discapacidad máxima en un plazo de dos semanas11. En enfermos que alcanzan la discapacidad máxima dentro de las 24 horas del inicio de la enfermedad o después de 4 semanas, deben considerarse diagnósticos alternativos23. El SGB tiene un curso clínico monofásico, aunque las fluctuaciones relacionadas con el tratamiento y las recaídas ocurren en una minoría de casos12,13.
Presentación clínica atípica
El SGB también puede presentarse de una manera atípi ca. Los signos de debilidad y sensitivos, si bien siempre son bilaterales, pueden ser asimétricos o predominan temente proximales o distales, y pueden comenzar en las piernas, los brazos o simultáneamente en todas las extremidades6,27. Además, el dolor intenso y difuso o la disfunción aislada del nervio craneal pueden preceder al inicio de la debilidad27. Los niños prescolares (< 6 años) especialmente pueden presentar características clínicas no específicas o atípicas, como dolor mal localizado, negativa a soportar peso, irritabilidad, meningismo o marcha inestable28,29. Si no se reconocen estos signos como una presentación temprana del SGB, es posible que el diagnóstico se retrase29. En una minoría de pacientes con SGB atípico, particularmente en aquellos que solo presentan signos motores (variante motora pura) y un subtipo de neuropatía axonal motora aguda en el examen electrofisiológico, se pueden observar reflejos normales o incluso exagerados durante el curso de la enfermedad30.
Variantes
Algunos pacientes tienen una variante clínica distinta y persistente de SGB que no se desarrolla de acuerdo con el patrón clásico de pérdida sensitiva y debilidad. Estas variantes incluyen debilidad sin signos sensitivos (variante motora pura); debilidad limitada a los nervios craneales (parálisis facial bilateral con parestesias), debilidad en miembros superiores (debilidad faríngea-cérvico-braquial) o miembros inferiores (variante paraparética); y el síndrome de Miller Fisher (MFS por sus siglas en inglés), que en su manifestación completa consiste en oftalmoplejía, arreflexia y ataxia (Fig. 2 y Tabla 1)6,31,32. En general, son escasas las veces en que las variantes de SGB son “puras” y a menudo se superponen en parte con el síndrome clásico o muestran características que son típicas de otras variantes33.
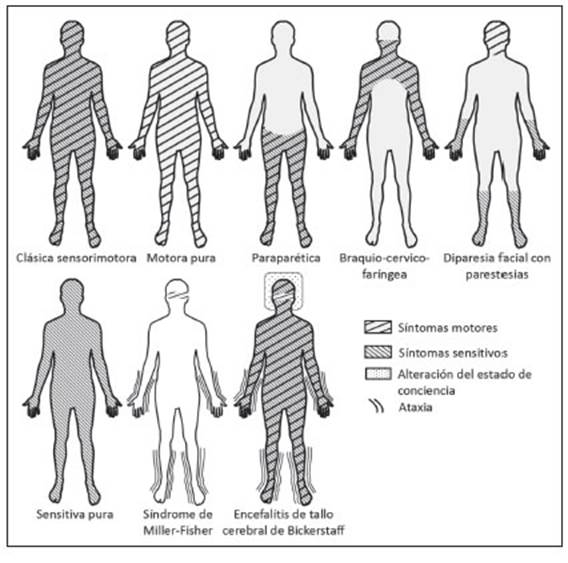

Además de las variantes enumeradas anteriormente, la ataxia sensitiva pura, la encefalitis de tallo cerebral de Bickerstaff (BBE por sus siglas en inglés) y una variante sensitiva pura, a menudo se incluyen en el espectro de SGB porque comparten características clínicas o fisiopatológicas con SGB. Sin embargo, la inclusión de estas variantes clíni cas está sujeta a debate ya que no cumplen con los criterios diagnósticos para el SGB (Anexo 1)2,3,32. La variante sen sitiva pura comparte características clínicas con la forma sensitivo-motora clásica del SGB, con la excepción de la presencia de síntomas y signos motores32,34; la ataxia sen sitiva pura y el MFS tienen perfiles clínicos superpuestos; y los pacientes con BBE suelen presentar síntomas que se asemejan a MFS y posteriormente desarrollan signos de disfunción del tallo cerebral, que incluyen deterioro de la conciencia y signos del tracto piramidal31-33,35-37. Al igual que los pacientes con MFS, los individuos con ataxia sensitiva o BBE pueden tener en su suero anticuerpos IgG contra GQ1b u otros gangliósidos31,35. Sin embargo, si el SGB sensitivo puro, la ataxia sensitiva pura y el BBE son variantes del SGB y/o una forma incompleta del MFS está sujeta a debate, y se requiere una evaluación diag nóstica detallada cuando se sospechan estas variantes (Anexos 1 y 2)32,34,36.
Eventos desencadenantes
Aproximadamente dos tercios de los pacientes que desa rrollan SGB informan síntomas de una infección dentro de las 6 semanas anteriores al inicio de la enfermedad11. Se cree que estas infecciones desencadenan la respuesta inmunitaria que causa el SGB 6. Seis patógenos se han asociado temporalmente con SGB en estudios de ca sos y controles: Campylobacter jejuni, citomegalovirus, virus de la hepatitis E, Mycoplasma pneumoniae, virus de Epstein-Barr y virus del Zika18,20,38. Se han sugerido otros patógenos relacionados con el SGB con base en pruebas microbiológicas de series de casos o estudios epidemiológicos, pero su papel en la patogénesis del SGB es incierto39-44. En general, la ausencia de una enfermedad anterior no excluye un diagnóstico de SGB, ya que las supuestas infecciones u otros estímulos inmunológicos pueden ser subclínicos.
Las vacunas se vincularon por primera vez al SGB en 1976 cuando se observó un aumento de 7.3 veces en el riesgo de SGB entre la población de los EE. UU. que ha bían recibido la vacuna contra la gripe “porcina”45. Desde entonces, el vínculo epidemiológico entre otras vacunas y el SGB se ha examinado en múltiples oportunidades, pero solo dos estudios adicionales han demostrado una relación entre el SGB y las vacunas contra la influenza46,47. Estos estudios sugirieron un aumento de aproxima damente un caso adicional de SGB por cada millón de vacunaciones, que es mucho menor que el observado para la vacuna contra la influenza de 197648,49. Ninguna otra vacuna se ha relacionado de manera convincente con el SGB15.
Se ha informado que existe una relación entre la ad ministración de medicamentos biológicos (por ejemplo, antagonistas del factor de necrosis tumoral, inhibidores de puntos de control inmunitarios o interferones de tipo I) y el SGB sobre la base de información de serie de casos y plausibilidad biológica50. Otros eventos -entre los que se incluyen la cirugía y las neoplasias- han sido relacio nados temporalmente con el SGB, pero estas relaciones carecen de una justificación biológica clara y la evidencia epidemiológica es limitada51,52.
Paso 2: ¿Cómo diagnosticar síndrome de Guillain-Barré?
En ausencia de biomarcadores de enfermedad suficien temente sensibles y específicos, el diagnóstico de SGB se basa en la historia clínica y el examen neurológico, y está respaldado por investigaciones complementarias, como el examen del LCR y los estudios de electro diagnóstico. Los dos grupos de criterios diagnósticos utilizados con mayor frecuencia para el SGB fueron desarrollados por el Instituto Nacional de Enfermeda des Neurológicas y Accidentes Cerebrovasculares de EE.UU. [National Institute of Neurological Disorders and Stroke] (NINDS) en 1978 (revisado en 1990) (Anexo 1)2,3 y por Brighton Collaboration en 2011 (*Tabla comple mentaria 1)2-4 Ambos grupos de criterios se diseñaron para investigar la asociación epidemiológica entre el SGB y las vacunas, pero desde entonces se han utilizado en otros estudios y ensayos clínicos. Consideramos que los criterios de NINDS son los más adecuados para uso clínico, ya que presentan las características clínicas de las formas típicas y atípicas del SGB, aunque los criterios de Brighton Collaboration también son importantes, se utilizan ampliamente y pueden ayudar a los médicos a clasificar los casos con SGB (típico) o MFS según la certeza del diagnóstico. También deben tenerse en cuenta varios diagnósticos diferenciales cuando se sospecha SGB, y algunos síntomas deben suscitar la sospecha de diagnósticos alternativos (Anexos 1 y 2). En la siguiente sección se describe con más detalle el papel de las investigaciones complementarias para confirmar un diagnóstico de SGB.
Estudios de laboratorio
Las pruebas de laboratorio se guían por el diagnóstico diferencial en pacientes individuales, pero en general, todos los pacientes con sospecha de SGB deberán tener hemogramas completos y análisis de sangre para gluco sa, electrolitos, función renal y enzimas hepáticas. Los resultados de estas pruebas pueden utilizarse para excluir otras causas de parálisis flácida aguda, como infecciones o disfunciones metabólicas o electrolíticas (Anexo 2). Se pueden realizar más pruebas específicas con el objetivo de descartar otras enfermedades que puedan simular al SGB (Anexo 2). Las pruebas para detectar infecciones previas no suelen contribuir al diagnóstico de SGB, pero pueden proporcionar información epidemiológica impor tante durante los brotes de enfermedades infecciosas, como se vio en brotes anteriores de infección por el virus del Zika y C. jejuni19,53 El valor diagnóstico de medir los niveles séricos de anticuerpos antigangliósidos es limi tado y es ensayo dependiente. Un resultado positivo de la prueba puede ser útil, especialmente cuando el diagnóstico está en duda, pero un resultado negativo de la prueba no descarta el SGB54. Los anticuerpos anti-GQ1b se encuentran en hasta en el 90% de los pacientes con MFS17, 55 y; por lo tanto, tienen mayor valor diagnóstico en pacientes con sospecha de MFS que en pacientes con SGB clásico u otras variantes. Cuando se sospecha de SGB, recomendamos no esperar los resultados de la prue ba de anticuerpos antes de comenzar con el tratamiento.
Examen de líquido cefalorraquídeo
El examen de LCR se utiliza principalmente para descartar causas de debilidad distintas del SGB y debe realizarse durante la evaluación inicial del paciente. El hallazgo clásico en el SGB es la combinación en el LCR, de un nivel elevado de proteínas y un recuento de células normal (conocido como disociación albúmino-citológica)56. Sin embargo, los niveles de proteínas son normales entre el 30 y el 50% de los pacientes en la primera semana posterior al inicio de la enfermedad y entre el 10 y el 30% de los pacientes en la segunda semana10,11,25,57. Por lo tanto, la concentración normal de proteína en el LCR no descarta el diagnóstico de SGB. La pleocitosis marcada (>50 células μl-1) sugiere otras patologías, como neoplasia leptomeníngea o enfer medades infecciosas o inflamatorias de la médula espinal o de las raíces nerviosas. Si bien la pleocitosis leve (10-50 células μl-1) es compatible con el SGB, debería impulsar a los médicos a considerar diagnósticos alternativos, como las causas infecciosas de polirradiculitis (Anexo 2)10,11.
Estudios electrodiagnósticos
No se requieren estudios electrodiagnósticos para diag nosticar el SGB. Sin embargo, recomendamos que estos estudios se realicen siempre que sea posible, ya que son útiles para respaldar el diagnóstico, especialmente en pacientes con una presentación atípica. En general, el examen electrofisiológico en pacientes con SGB revelará una polirradiculoneuropatía o polineuropatía sensitivo-motora, indicada por velocidades de conducción reduci das, reducción de las amplitudes motoras y sensitivas, dispersión temporal anormal y/o bloqueos parciales de conducción motora6,58. El SGB presenta típicamente un “patrón de preservación sural” en el que el potencial de acción del nervio sensitivo sural es normal, mientras que los potenciales de acción de los nervios sensitivos medianos y cubitales son anormales o incluso están ausentes6,58. Sin embargo, los estudios electrofisiológicos pueden ser normales cuando se realizan temprano en el curso de la enfermedad (dentro de la semana del inicio de los síntomas) o en pacientes con debilidad inicialmente proximal, enfermedad leve, progresión lenta o variantes clínicas5,59,60. En estos pacientes, puede ser útil repetir el estudio electrodiagnóstico dos a tres semanas después. En pacientes con MFS, los resultados de los estudios electrodiagnósticos suelen ser normales o demostrar solo una amplitud reducida de los potenciales de acción de los nervios sensitivos4,61.
Los estudios electrodiagnósticos también pueden diferenciar entre los tres subtipos electrofisiológicos de SGB clásico: AIDP, AMAN, AMSAN. Existen varios grupos de criterios electrodiagnósticos que tienen como objetivo clasificar a los pacientes en estos diferentes subtipos electrofisiológicos sobre la base de la presencia de características específicas electrodiagnósticas en, al menos, dos nervios motores. Aún no se ha alcanzado un consenso internacional sobre qué grupo de criterios define mejor los subtipos electrofisiológicos5,53,62. Sin embargo, alrededor de un tercio de los pacientes con SGB no cumplen con ninguno de estos criterios y son etiquetados como “equívocos” o “inexcitables”. Algunos estudios han demostrado que la repetición de los estudios electrodiagnósticos entre 3 y 8 semanas posteriores al inicio de la enfermedad podría ayudar a la clasificación electrodiagnóstica, al permitir la clasificación de casos que inicialmente no eran clasificables, o la reclasificación de casos que inicialmente se clasificaron como AIDP, AMAN o AMSAN, aunque esta práctica es controvertida63-65.
Neuroimágenes
La resonancia magnética no forma parte de la evaluación diagnóstica de rutina del SGB, pero puede ser útil, espe cialmente para excluir diagnósticos diferenciales como infección del tallo cerebral, accidente cerebrovascular, inflamación de la médula espinal o de las células del asta anterior de la médula, compresión de la raíz nerviosa o neoplasia leptomeníngea (Anexo 2). La presencia de real ce de la raíz nerviosa en la resonancia magnética realzada con gadolinio es una característica no específica pero sugestiva del SGB66 y puede respaldar un diagnóstico de SGB, especialmente en niños pequeños, en quienes la evaluación clínica y electrofisiológica puede ser muy difícil67. A la luz de los recientes brotes de mielitis flácida aguda en niños pequeños, cuya presentación clínica puede simular el SGB, se debe prestar especial atención al uso de la resonancia magnética para distinguir entre estos dos diagnósticos68, 69. Sin embargo, los médicos deben tener en cuenta que el realce de la raíz nerviosa se puede encontrar en una minoría de personas con mielitis flácida aguda70.
Una nueva herramienta de diagnóstico útil en el SGB son las imágenes por ultrasonido de los nervios periféri cos, que ha revelado raíces nerviosas cervicales hipertró ficas al principio del curso de la enfermedad, lo que indica la importancia de la inflamación de la raíz espinal como un mecanismo patológico temprano71, 72. Por lo tanto, esta técnica podría ayudar a establecer un diagnóstico de GBS temprano en el curso de la enfermedad, aunque se requiere una mayor validación.
Paso 3: ¿Cuándo ingresar a la Unidad de Cuidados Intensivos?
Entre los criterios de admisión a la unidad de cuidados intensivos (UCI) se incluyen las siguientes: dificultad respiratoria progresiva con insuficiencia respiratoria inminente, disfunción cardiovascular disautonómica grave (por ejemplo, arritmias o variación marcada en la presión arterial), trastorno grave de la deglución o disminución del reflejo de la tos, y progresión rápida de la debilidad73,74. Un estado de insuficiencia respiratoria inminente se define como signos clínicos de dificultad respiratoria, que incluyen disnea en reposo o al hablar, incapacidad para contar hasta 15 en una sola respiración, uso de los mús culos respiratorios accesorios, aumento de la frecuencia respiratoria o cardíaca, capacidad vital de < 15-20 ml/kg o < 1 l, o mediciones anormales de gases en sangre arterial u oximetría de pulso.
Dado que hasta el 22% de los pacientes con GBS re quieren ventilación mecánica dentro de la primera semana de ingreso, los enfermos con riesgo de falla respiratoria de ben ser identificados lo antes posible75. La herramienta de pronóstico de puntaje de insuficiencia respiratoria Erasmus del SGB [Erasmus GBS Respiratory Insufficiency Score] (EGRIS) https://gbstools.erasmusmc.nl/prognosis-tool, se desarrolló para este propósito y calcula la probabilidad (1- 90%) de que un paciente requiera ventilación dentro de una semana a partir de la evaluación (Anexo 3)75.
Los factores de riesgo para la ventilación mecánica prolongada incluyen la incapacidad para levantar los brazos de la cama una semana después de la intuba ción y el subtipo axonal, o nervios no excitables en los estudios electrofisiológicos76. Se debe considerar la traqueostomía temprana en pacientes que tienen estos factores de riesgo.
Paso 4: ¿Cuándo iniciar el tratamiento?
Se debe iniciar la terapia inmunomoduladora si los pa cientes no pueden caminar de forma independiente 10 metros77,78. La evidencia sobre la eficacia del tratamiento en pacientes que aún pueden caminar de forma indepen diente es limitada, pero se debe considerar el tratamiento especialmente si estos pacientes presentan debilidad rápidamente progresiva u otros síntomas graves como disfunción autonómica, insuficiencia bulbar o insuficiencia respiratoria79-81. Los ensayos clínicos han demostrado un efecto de tratamiento para la inmunoglobulina intravenosa (IgIV) cuando se inicia dentro de las 2 semanas posterio res al inicio de la debilidad y para la plasmaféresis cuando se inicia dentro de las 4 semanas77,78. No hay pruebas de eficacia más allá de estos períodos de tiempo.
Paso 5: Opciones de tratamiento
Estrategias de tratamiento
La IgIV (0.4 g por kg de peso corporal diariamente durante 5 días, equivalente a 2 gramos por kilo dosis total) y la plasmaféresis (200-250 ml de plasma por kg de peso corporal en cinco sesiones) son tratamientos igualmente eficaces para el SGB77,81. La IgIV y la plasmaféresis con llevan riesgos comparables de eventos adversos, aunque los primeros estudios mostraron que la plasmaféresis tenía más probabilidades de interrumpirse que la IgIV77,82. Dado que la IgIV también es más fácil de administrar y, en gene ral, tiene una mayor disponibilidad que la plasmaféresis, suele ser el tratamiento de elección. Además de la IgIV y la plasmaféresis, ningún otro procedimiento o fármaco ha demostrado ser eficaz en el tratamiento del SGB. Si bien podría esperarse que los corticosteroides fueran beneficio sos para reducir la inflamación y, por lo tanto, la progresión de la enfermedad en el SGB, ocho ensayos controlados aleatorizados sobre la eficacia de los corticosteroides para el SGB no mostraron un beneficio significativo, e incluso se demostró que el tratamiento con corticosteroides orales presenta resultados negativos83. Además, la plasmaféresis seguida de IgIV no es más eficaz que cualquiera de los tratamientos independientes y no se dispone de pruebas suficientes de la eficacia del tratamiento complementario con metilprednisolona intravenosa en pacientes tratados con IgIV83,84. En entornos clínicos donde los recursos son limitados, la plasmaféresis en pequeño volumen podría ser una alternativa económica y relativamente segura ante la plasmaféresis convencional, pero este enfoque no puede recomendarse para uso general hasta que se haya esta blecido su eficacia en ensayos adicionales85.
Se puede considerar el tratamiento antimicrobiano o antiviral en pacientes con SGB que tengan una infección en curso; sin embargo, las infecciones anteriores suelen resolverse antes del inicio de la debilidad.
Grupos específicos de pacientes
Variantes del síndrome de Guillain-Barré
Los pacientes con MFS puro tienden a tener un curso de la enfermedad relativamente leve y la mayoría se recupera completamente sin tratamiento dentro de los 6 meses86. Por lo tanto, el tratamiento generalmente no se recomienda en este grupo de pacientes, pero ellos deben ser cuidadosamente controlados porque un sub grupo puede desarrollar debilidad en las extremidades, parálisis bulbar o facial o insuficiencia respiratoria33,81. La gravedad de la evolución de la enfermedad de la BBE justifica el tratamiento con IgIV o plasmaféresis, aunque la evidencia de la eficacia del tratamiento en este contexto es limitada35,86. Para las otras variantes clínicas, actualmente no hay evidencia con respecto al tratamiento, aunque muchos expertos administrarán IgIV o plasmaféresis87.
Embarazadas
Ni la IgIV ni la plasmaféresis están contraindicadas du rante el embarazo. Sin embargo, como la plasmaféresis requiere consideraciones y seguimiento adicionales, se podría preferir la IgIV88-90.
Niños
No hay indicios de que sea necesario desviarse de la prác tica estándar de los adultos al tratar a niños con SGB77,79,91. La evidencia sobre la eficacia relativa de la plas maféresis y la IgIV en niños es limitada91. Sin embargo, como la plasmaféresis solo está disponible en centros con experiencia en su uso y parece producir mayor malestar y mayores tasas de complicaciones que la IgIV en niños, la IgIV suele ser la terapia de primera línea para niños con SGB92. Aunque algunos centros pediátricos administran IgIV, 2 g/kg (peso corporal) durante 2 días, en lugar del régimen estándar para adultos de 2 g/kg (peso corporal) durante 5 días, un estudio indicó que la fluctuación relacionada con el tratamiento era más frecuente con un régimen de 2 días (5 de 23 niños) que con el régimen de 5 días (0 de 23 niños)79.
Paso 6: Seguimiento de la progresión de la enfermedad
Se requiere una evaluación regular para monitorear la pro gresión de la enfermedad y la aparición de complicaciones. En primer lugar, se recomienda la medición rutinaria de la función respiratoria, ya que no todos los pacientes con insuficiencia respiratoria tendrán disnea. La evaluación respiratorias incluye el uso de los músculos respiratorios accesorios, conteo durante la espiración de una respira ción inspiratoria a plena capacidad (una sola respiración de ≤ 19 predice la necesidad de ventilación mecánica), la capacidad vital y la presión inspiratoria y espiratoria máxi ma74,93. Los médicos deben considerar el uso de la ‘regla 20/30/40’, según la cual se considera al paciente en riesgo de insuficiencia respiratoria si la capacidad vital es < 20 ml/kg, la presión inspiratoria máxima es < 30 cm H2O o la presión espiratoria máxima es < 40 cm H2O94. En segun do lugar, la fuerza muscular en el cuello, los brazos y las piernas debe evaluarse mediante la escala de calificación del Medical Research Council https://gbstools.erasmusmc.nl/prognosis-tool, o una escala similar, y la discapacidad funcional debe evaluarse en la escala de discapacidad SGB (*Tabla complementaria 2), una herramienta ampliamente utilizada para documentar el curso de la enfermedad del SGB95. En tercer lugar, se debe controlar a los pacientes a fin de detectar dificultades para tragar y toser. Por último, la disfunción autonómica debe evaluarse mediante elec trocardiografía y monitoreo de la frecuencia cardíaca, la presión arterial y la función intestinal y vesical.
La naturaleza y la frecuencia del seguimiento depen den de la tasa de deterioro, la presencia o ausencia de disfunción autonómica, la fase de la enfermedad y el entorno sanitario, y deben evaluarse cuidadosamente en cada paciente individual. Hasta dos tercios de las muertes de pacientes con SGB ocurren durante la fase de recuperación y son causadas principalmente por dis función cardiovascular y respiratoria6,7,11. Por lo tanto, recomendamos a los médicos que se mantengan alerta durante esta fase y controlen al paciente para detectar posibles arritmias, cambios de presión arterial o dificultad respiratoria causada por tapones mucosos. Este monito reo es especialmente importante en pacientes que han salido recientemente de la UCI y en aquellos pacientes con factores de riesgo cardiovascular.
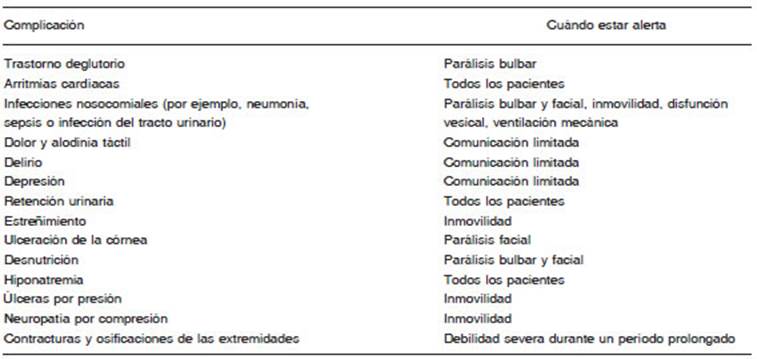
Paso 7: Tratamiento de complicaciones tempranas
Las complicaciones en los pacientes con SGB pueden ocasionar morbilidad grave y muerte96. Algunas de es tas complicaciones, incluidas las úlceras por presión, las infecciones nosocomiales (por ejemplo, neumonía o infecciones del tracto urinario) y la trombosis venosa profunda, pueden ocurrir en cualquier paciente hospi talizado postrado en una cama, y se recomiendan las medidas preventivas y el tratamiento de práctica estándar. Existen otras complicaciones más específicas del SGB, por ejemplo, la incapacidad para tragar con seguridad en pacientes con parálisis bulbar; ulceración de la córnea en pacientes con parálisis facial; y contracturas de las extre midades, osificación y parálisis por presión en pacientes con debilidad en las extremidades (Tabla 2). El dolor, las alucinaciones, la ansiedad y la depresión también son frecuentes en el SGB, y los cuidadores deben preguntar específicamente a los pacientes si están experimentando estos síntomas, especialmente si los pacientes tienen capacidades limitadas de comunicación y/o están en la UCI. El reconocimiento y el tratamiento adecuado de los síntomas psicológicos y el dolor en una etapa temprana es importante porque estos síntomas pueden tener un efecto importante en el bienestar de los pacientes. Los cuidadores también deben ser conscientes de que los pa cientes con SGB, incluso aquellos con parálisis completa, generalmente tienen la conciencia, la visión y la audición intactas. Por lo tanto, es importante tener en cuenta lo que se dice junto a la cama del paciente y explicarles la naturaleza de los procedimientos para reducir la ansiedad. El manejo adecuado de las complicaciones resulta más eficaz si lo lleva a cabo un equipo multidisciplinario, que puede incluir enfermeros/as, fisioterapeutas, especialistas en rehabilitación, terapeutas ocupacionales, fonoaudió logos y nutricionistas.
Paso 8: Tratamiento de la progresión clínica
Respuesta insuficiente al tratamiento
Aproximadamente el 40% de los pacientes tratados con dosis estándar de plasmaféresis o IgIV no mejoran en las primeras 4 semanas posteriores al tratamiento81,83. Tal progresión de la enfermedad no implica que el tratamiento sea ineficaz, ya que la progresión podría haber sido peor sin terapia6. Los médicos pueden considerar repetir el tratamiento o cambiar a un tratamiento alternativo, pero en la actualidad no existe evidencia de que este enfoque mejore el resultado97,98. Existe un ensayo clínico en curso que investiga el efecto de administrar una segunda dosis de IgIV99.
Fluctuaciones relacionadas con el tratamiento
Los fluctuaciones relacionada con el tratamiento se observan en el 6-10% de los pacientes con SGB y se definen como la progresión de la enfermedad que ocurre dentro de los 2 meses posteriores a una mejoría o es tabilización clínica inicial inducida por el tratamiento12,13. Las fluctuaciones relacionadas con el tratamiento deben distinguirse de la progresión clínica sin ninguna respues ta inicial al tratamiento. La opinión general es que una fluctuación relacionada con el tratamiento indica que el efecto del tratamiento ha desaparecido mientras la fase inflamatoria de la enfermedad aún está en curso. Por lo tanto, los pacientes con SGB que muestran fluctuación relacionada con el tratamiento podrían beneficiarse con un tratamiento adicional, y repetir el ciclo completo de IgIV, o recambio plasmático, que en estos pacientes es una práctica común, aunque faltan pruebas que respalden este enfoque81.
Polineuropatía desmielinizante inflamatoria crónica
En ~ 5% de los pacientes con SGB, las recaídas clíni cas repetidas sugieren un proceso de enfermedad más crónico y el diagnóstico se cambia a polineuropatía des mielinizante inflamatoria crónica de inicio agudo (CIDP)12. La CIDP de inicio agudo suele presentarse con tres o más fluctuaciones relacionadas con el tratamiento y/o deterioro clínico ≥ 8 semanas posteriores al inicio de la enfermedad12.
Paso 9: Predicción de resultados
La mayoría de los pacientes con SGB, incluso aquellos que estaban tetrapléjicos en el nadir o que requirieron ventilación mecánica durante un período prolongado, muestran una recuperación extensa, especialmente en el primer año después del inicio de la enfermedad11,100. Aproximadamente el 80% de los pacientes con SGB recuperan la capacidad para caminar de forma independiente a los 6 meses del inicio de la enfermedad11. La probabilidad de recuperar la capacidad para caminar se puede calcular en cada paciente a través de la herramienta de pronóstico de puntaje de resultado Erasmus GBS modificado (mEGOS) https://gbstools.erasmusmc.nl/prognosis-tool., (*Tabla complementaria 3)101.
A pesar de las perspectivas generalmente positivas para los pacientes con SGB, la muerte ocurre entre el 3 y el 10% de los casos, con mayor frecuencia debido a complicaciones cardiovasculares y respiratorias, que pueden ocurrir tanto en la fase aguda como en la de re cuperación7-9. Entre los factores de riesgo de mortalidad se incluyen la edad avanzada y enfermedad grave al inicio7. Las quejas residuales a largo plazo también son comunes y pueden incluir dolor neuropático, debilidad y fatiga102-104. Sin embargo, la recuperación de estas mo lestias aún puede ocurrir >5 años después del inicio de la enfermedad104.
Los episodios recurrentes de SGB son poco comunes y afectan entre el 2 y el 5% de los pacientes, pero este porcentaje sigue siendo mayor que el riesgo de por vida de padecer SGB en la población general (0.1%)14,15. Muchas vacunas tienen una advertencia sobre el SGB, aunque el SGB previo no es una contraindicación estricta para la vacunación. La discusión con expertos podría resultar de gran ayuda para los pacientes que fueron diagnosticados con SGB < 1 año antes de una vacunación planificada o que desarrollaron previamente SGB poco después de recibir la misma vacuna. En estos casos, es necesario so pesar los beneficios de la vacunación para enfermedades específicas (por ejemplo, influenza en personas de edad avanzada) frente al riesgo pequeño y posiblemente solo teórico de un episodio recurrente de SGB14.
Paso 10: Planificación de la rehabilitación
Los pacientes con SGB pueden experimentar una varie dad de problemas residuales a largo plazo, incluida la recuperación incompleta de la función motora y sensorial, así como fatiga, dolor y distrés psicológico104. Antes de que el paciente sea dado de alta, se deben considerar y tratar estos posibles efectos a largo plazo del SGB105,106.
Condición física
La organización de un programa de rehabilitación con un especialista en rehabilitación, fisioterapeuta y terapeuta ocupacional es un paso crucial hacia la recuperación. Los programas deben apuntar a reducir la discapacidad en las primeras etapas de la recuperación y luego a restaurar la función motora y sensorial y la condición física a los niveles previos a la enfermedad107. Se ha demostrado que los programas de ejercicio para pacientes con SGB, que incluyen ejercicios de amplitud de movimiento, ciclismo estacionario, entrenamiento de fuerza y caminata, me joran la condición física, la capacidad para caminar y la independencia en las actividades de la vida diaria107. Sin embargo, la intensidad del ejercicio debe controlarse de cerca ya que el exceso de esfuerzo puede causar fatiga107.
Fatiga
La fatiga que no está relacionada con los déficits motores residuales se encuentra en el 60 a 80% de los pacien tes con SGB y suele ser una de las expresiones más incapacitantes108,109. Se deben considerar otras causas antes de concluir que la fatiga en un paciente es un pro blema residual de SGB. Al igual que con la recuperación de la función física, se ha demostrado que un programa de ejercicio supervisado y escalonado resulta útil para reducir la fatiga110.
Dolor
Al menos un tercio de los pacientes con SGB informan de dolor intenso un año después del inicio de la enfermedad. Este dolor puede persistir durante > 10 años14,27. El dolor crónico en el SGB se caracteriza por dolor muscular en la zona lumbar y las extremidades, parestesias dolorosas, artralgia y dolor radicular. Aunque la patogénesis de este dolor no se comprende completamente, el dolor muscular y la artralgia pueden ser atribuibles a la inmovilidad, y el dolor neuropático puede ser causado por la regeneración o el daño persistente de las fibras nerviosas pequeñas27. Entre las estrategias de tratamiento se incluyen fomen tar la movilización y administrar fármacos para el dolor neuropático o nociceptivo105.
Consecuencias psicológicas
A menudo en individuos previamente sanos, la pérdida rápida de la función física puede ser muy traumática, causar ansiedad y/o depresión. El reconocimiento y el tratamiento tempranos del distrés psicológico es impor tante en pacientes con SGB, especialmente porque el estado mental puede influir en la recuperación física y viceversa110,111; la derivación a un psicólogo o psiquiatra puede ser beneficiosa para algunos pacientes111. Brindar información precisa a los pacientes sobre las posibilidades de recuperación relativamente buenas y el bajo riesgo de recurrencia (2-5%) puede ayudar a reducir su miedo11,14. Conectar a los pacientes con otras personas que han tenido GBS también puede ayudar a guiarlos a través del proceso de rehabilitación. GBS/CIDP Foundation Internationalhttps://www.gbs-cidp.org/ la asociación inter nacional de pacientes para GBS, y otras organizaciones nacionales pueden ayudar a establecer estas redes.
Discusión
El SGB puede ser una enfermedad de diagnóstico y manejo complejo, ya que la presentación clínica es heterogénea y el pronóstico varía ampliamente entre pacientes. El manejo del SGB puede resultar especialmente complicado durante los brotes provocados por enfermedades infecciosas, como se observó recientemente durante la epidemia por el virus Zika. En ausencia de una guía clínica internacional para el SGB, hemos preparado esta guía de consenso para el diagnóstico y el tratamiento del SGB, que fue desarrollada por un equipo de neurólogos clínicos de todo el mundo y está diseñada para aplicarse a nivel general en todos los entornos clínicos, independientemente de las capacidades de los especialistas o la disponibilidad de los recursos. El diseño paso a paso se utilizó para centrar la atención en los problemas más importantes del SGB y para facilitar el uso de la guía en la práctica clínica.
A medida que se desarrolle el campo de la investiga ción del GBS y los estudios en curso tengan como objetivo mejorar el diagnóstico, el tratamiento y el modelado de pronóstico, esta guía deberá actualizarse periódicamente. Por ejemplo, la ecografía de los nervios periféricos emerge como una herramienta de diagnóstico potencial y podría requerir más comentarios en versiones futuras de estos lineamientos. En relación con el tratamiento, se está in vestigando la eficacia de los inhibidores del complemento, las enzimas que segmentan IgG y un segundo ciclo de IgIV78,112,113. Hay poca información sobre cómo medir y predecir el resultado a largo plazo en pacientes con SGB, y se necesitan estudios de validación de modelos de pronósticos conocidos (por ejemplo, mEGOS y EGRIS) e investigación de nuevas medidas de resultado. Tenemos la intención de buscar comentarios sobre esta guía y proporcionar actualizaciones basadas en los resultados de estudios en curso e investigaciones futuras.
Para mejorar aún más la gestión mundial del SGB, nuestro objetivo es utilizar este informe de consenso como base para el desarrollo de recursos de información en línea, material de capacitación y cursos de enseñanza. Estos recursos se dirigirán a los trabajadores de la salud, incluidos los neurólogos clínicos, así como a los pacientes con SGB y sus familiares.
Nota: Información suplementaria: https://doi.org/10.1038/s41582-019-0250-9.


