











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkLa leucemia de células grandes granulares (LGLL) es una enfermedad caracterizada por infiltración linfocítica de sangre periférica y médula con células grandes granu lares, esplenomegalia y citopenias, siendo la neutropenia la más común. Contribuye del 2 al 5% de los trastornos linfoproliferativos crónicos en Norte América1. La edad media de aparición es de 60 años y no tiene predilección por tipo de sexo2.
Normalmente, los LGL corresponden entre el 10 y el 15% de las células mononucleares de sangre periférica y se dividen en 2 linajes mayores, células T CD3 + y células natural killer. Los recuentos de estas células circulantes son en promedio 0.25 × 109/l, pero la mayoría de los pa cientes con este tipo de leucemia oscilan con exceso de entre 2 × 109 y 10 × 109/l3,4.
La leucemia de células T grandes granulares (LGLL-T) es el tipo más común de LGLL, representa aproximada mente el 85% de los pacientes5. El diagnóstico se hace con conteo de LGL en sangre periférica > 500 mm3 y con una inmunofenotipificación por citometría de flujo. Los criterios específicos para la LGLL-T incluyen: expresión de marcadores de superficie LGL compatibles con un fenotipo de células T activadas (comúnmente CD3 + / CD8 + / CD57 + y / o CD16 +) y reordenamiento clonal del gen TCR-γ usando PCR o expresión de Vβ específica y clonal usando FCM4.
Su curso suele ser indolente y presentarse con leu copenia o linfocitosis. Se asocia con artritis reumatoide (AR) (11-36%) y neoplasias malignas de células B (5-7%) y con menor frecuencia a otras enfermedades autoinmu nes como el síndrome de Sjögren, enfermedad celíaca, hipertensión pulmonar, y trastornos hematológicos como el síndrome mielodisplásico y trasplante de células he matopoyéticas6.
La presentación clínica más común es la neutrope nia (aproximadamente el 85% de los casos) a menudo acompañada de infecciones o neutropenia febril. Sin embargo, a diferencia de la neutropenia asociada con otros trastornos hematológicos, los pacientes con leuce mia de células grandes granulares pueden permanecer sorprendentemente libres de complicaciones infecciosas durante períodos prolongados6.
En cuanto a las citopenias de origen autoinmune puede encontrarse aplasia pura de células rojas, anemia hemolítica autoinmune, neutropenia y trombocitopenia autoinmune. Sin embargo, el síndrome de Evans asociado a la LGLL es infrecuente7.
En esta ocasión presentamos una paciente con ante cedente de LGLL-T indolente y sospecha de AR que se presenta a nuestro servicio con un síndrome de Evans.
Caso clínico
Mujer de 72 años con antecedente de LGLL-T diagnosticada en diciembre de 2019 en contexto de linfomonocitosis crónica, con recuento leucocitario de 7836/mm3, 29.9% de neutrófilos segmentados, 49.2% de linfocitos y 20.57% de monocitos (Tabla 1). Para arribar al diagnóstico, se realizó frotis de sangre periférica que mostró un 60% de linfocitos predominantemente grandes granulares con abundante citoplasma con gránulos azurófilos, citometría de flujo de sangre periférica que de tectó un 16.3 % de linfocitos T CD3+ CD4+ CD8- maduros con perfil de marcación aberrante, es decir, que presentó un fenotipo distinto cuando se comparó con la población normal remanente, debido a la sobreexpresión de CD5 y CD7, y evaluación de clonalidad T que detectó una población con patrón de reordenamiento monoclonal bialélico en el gen del receptor de linfocitos T (TCR) (Fig. 1). No presentó criterios de tratamiento en ese momento.

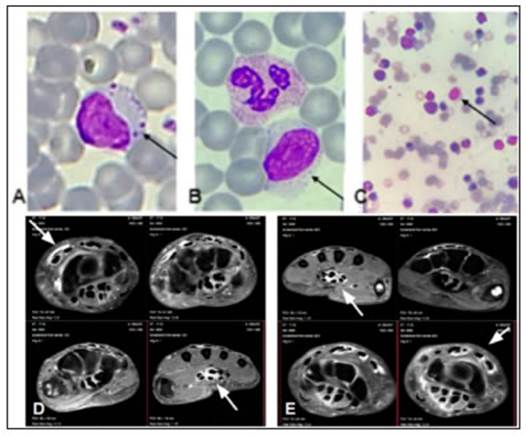
Fig. 1 A y B) Frotis de sangre periférica observados por microscopio óptico y tinción de hematoxilina y eosina (imagen ampliada de 100X). En la imagen A se observa un lin focito de gran tamaño con ligera expansión del citoplasma y la presencia de gránulos prominentes azurófilos (flecha). El núcleo es excéntrico y presenta cromatina madura. En el fondo se pueden observar hematíes y plaquetas. En la imagen B se observa un linfocito grande granular con núcleo en posición central y notoria expansión del citoplasma, junto a un neutrófilo (flecha). C) Medulograma de aspirado de médula ósea teñido con hematoxilina y eosina (microscopio óptico, 100x). Se señala la presencia de un linfocito grande granular junto a elementos mieloides y eritroides (flecha). D y E) Resonancia magnética de mano izquierda (D) y de mano derecha (E) sin contraste (secuencia T2 SPAIR): tenosinovitis de los flexores profundos y superficiales de los dedos, y de los compartimentos extensores (flechas)
Consultó a nuestro centro a principios de marzo de 2021 por disnea, fatiga, astenia, adinamia y registros febriles ves pertinos de aproximadamente dos meses de evolución. Al interrogatorio dirigido refirió pérdida de peso de aproximada mente 10 kilos en los últimos dos meses, artralgias y artritis de 1 año de evolución, bilateral, simétrica, con compromiso de pequeñas y grandes articulaciones a predominio de manos, acompañado de rigidez matinal de 1 hora de duración. En el laboratorio de ingreso se destacó hemoglobina de 4.4 g/dl, hematocrito 12.8%, leucocitos totales 16 000/mm3 (neutrófilos segmentados 64.4%, linfocitos 12.5%, y monocitos 22.5%), plaquetas 9000/mm3, recuento de reticulocitos 206 000/mm3 (18.8%), LDH 466 UI/l, bilirrubina 3.9 mg/dl a predominio indi recto, y haptoglobina sérica menor a 5.83 mg/dl (Tabla 1). Se decidió internación para estudio y tratamiento. La prueba de Coombs directa fue positiva, a expensas de la presencia en la membrana eritrocitaria de inmunoglobulinas G. Se realizó tomografía de tórax y abdominopelviana con doble contraste que presentó como único hallazgo relevante esplenomegalia leve homogénea de 131mm. En relación con los antecedentes articulares, con un alto índice de sospecha de AR, y para descartar causas secundarias de síndrome de Evans, se solicitó prueba de látex que resultó positiva de 31.47 UI/ml (método: turbidimetría, referencia: negativo menor a 12.50), anticuerpos antipéptidos citrulinados (ACPA) negativos 6.9 CU (método quimioluminiscencia, referencia: negativo menor a 20), anticuerpos antinucleares 1/80 patrón citoplasmático granular fino (método: inmunofluorescencia indirecta, sustrato: HEp 2, dilución de corte en adultos: 1/80), anticuerpos anti Sm negativo (método: Enzimo Inmuno Ensayo), anti ds-ADN negativo (método: inmunofluorescencia indirecta, sustrato: Crithidia luciliae, dilución de corte: 1/10), anticoagulante lú pico negativo, anticardiolipina negativo, anti B2 glicoproteína negativo, antiRo y antiLA negativos, con C3 y C4 de 60 mg/ dl y 14 mg/dl respectivamente (referencia: 83-177 y 10-40), proteinograma con hipergammaglobulinemia policlonal 3.53 g/dl (referencia: 0,70-1,47), IgA sérica 92 mg/dl (referencia: 70-400), y orina completa sin particularidades. Como otras causas secundarias de anemia hemolítica y trombocitopenia autoinmune se descartaron hepatitis virales y HIV. La radio grafía de manos y pies no presentó evidencia de enfermedad erosiva ni osteopenia en banda, y la ecografía reumatológica no demostró actividad inflamatoria.
Se practicó una punción aspiración de médula ósea con anatomía patológica que demostró un aumento de celulari dad, hiperplasia de serie eritroide, cambios displásicos en la serie mieloide y megacariocítica, y 4% de linfocitos T con la misma marcación por citometría que la hallada previamente en sangre periférica (Fig. 1).
Se interpretaron estos resultados como un síndrome de Evans, secundario a LGLL-T, por lo que se inició mepredni sona a 1 mg/kg/día e inmunoglobulina humana 1 g/kg por 2 días. Durante las primeras 72 horas de internación se realizaron transfusiones de glóbulos rojos (5 unidades), y plaquetas (7 unidades), logrando mejoría sintomática. Debido a la gravedad del cuadro, el riesgo de recaída y pensando en acelerar el descenso progresivo de corticoides en el corto plazo, se decidió iniciar rituximab 375 mg/m2/dosis/semana por 4 semanas, a los 14 días de internación. Se otorgó el alta hospitalaria al día 15 de internación por buena evolución clínica y de laboratorio (Tabla 1).
A los 90 días de iniciado el cuadro, la paciente continuó mejorando los parámetros del laboratorio (Tabla 1). Se repitió la prueba de látex que resultó positiva de 18.82 UI/ml, con nuevos ACPA, anticuerpos antinucleares, anticuerpos anti Sm, y anti ds-ADN negativos. En contexto de descenso progresivo de meprednisona (2 mg/día), se exacerbó el compromiso arti cular con dolor, edema y rigidez matinal de 1 hora de duración, a predominio de manos, muñecas, rodillas, y tobillos. La pa ciente refería parestesias en ambas manos, con sospecha de síndrome del túnel carpiano. Se solicitó resonancia de ambas manos sin contraste, que demostró tenosinovitis bilateral de los flexores profundos y superficiales de los dedos, y de los compartimentos extensores, y edematización de los nervios medianos, interpretándose como AR asociada a LGLL-T (Fig. 1). Se indicó meprednisona 8 mg/día y metotrexato 15 mg por semana con buena respuesta.
Discusión
En esta comunicación describimos a una paciente con dos manifestaciones autoinmunes presentadas en un contexto atípico e infrecuente, asociadas a la LGLL-T.
La AR es la enfermedad asociada con mayor fre cuencia y se presenta en aproximadamente el 25% de los pacientes. El inicio de la AR en comparación con el de la LGLL-T es variable; la proliferación clonal puede preceder al desarrollo de AR por varios años, o diagnosticarse simultáneamente. Por el contrario, la proliferación de LGL es mucho menos común en pacientes con AR. Un estudio de 2018 de 529 pacientes con AR que aprovechó la mayor sensibi lidad de la citometría de flujo y los reordenamientos moleculares para detectar los LGL clonales describe una prevalencia del 3.6% en AR8,9. Los estudios de secuenciación de última generación han identificado mutaciones en el transductor de señal y activador del gen de la transcripción 3 (STAT3) en pacientes con LGLL. Aquellos con mutaciones STAT3 tienen una probabilidad significativamente mayor, en compara ción con los pacientes con LGLL sin las mutaciones, de tener AR y de presentar neutropenia (26 frente al 6% y 77 frente al 50%, respectivamente)10.
Existe una amplia gama de gravedad de la artritis, desde una inflamación articular leve e intermitente hasta una artropatía deformante progresiva11. La mayoría de los pacientes tienen pruebas positivas para el factor reumatoide y los anticuerpos antipéptidos citrulinados; ocasionalmente anticuerpos antinucleares11. Un estudio de más de 500 pacientes con AR no demostró diferencias clínicas entre pacientes con y sin LGL clonales excepto por su mayor exposición a terapia con inhibidores del factor de necrosis tumoral8.
El síndrome de Felty (SF), la tríada de artritis crónica, esplenomegalia y granulocitopenia, se presenta típica mente en pacientes con AR seropositiva grave de larga duración, a menudo en asociación con otras manifestacio nes extraarticulares. Muchos de ellos tienen proliferación de LGL, y algunos pacientes previamente diagnosticados con SF ahora se clasificarían como LGLL-T, utilizando técnicas de inmunofenotipificación y análisis molecu lares9,12. La distinción entre SF y LGLL-T en pacientes con AR generalmente se ha basado en la determinación de si la proliferación de LGL es policlonal o monoclonal, respectivamente. Sin embargo, esta distinción no siempre es clara. En pacientes con AR, el SF es la afección que con mayor probabilidad se confunde con la LGLL-T y, a veces, se considera parte de un espectro común.
Los pacientes con LGLL-T y AR suelen tener un curso crónico e indolente con una mediana de supervivencia de más de 10 años, aunque la mayoría eventualmente re quiere tratamiento13. Se recomienda el uso de metotrexato (10 mg/m2 por semana) con o sin prednisona para los pacientes con LGLL-T con un trastorno autoinmunitario asociado13.
En la paciente del caso presentado, el síndrome de Evans se interpretó como secundario a LGLL-T, luego de descartar otras causas tales como déficit de inmunoglobulina A, inmunodeficiencia común variable, epatitis virales, HIV y lupus eritematoso sistémico14,15. La poliartritis bilateral y simétrica, la tenosinovitis de ambas manos confirmada por resonancia magnética, y el factor reumatoideo positivo bajo, apoyaron el diagnóstico de AR temprana asociada a LGLL-T, cumpliendo con los criterios de clasificación de 2010 del American College of Rheu matology y la European League Against Rheumatism.

